天昊生物16S扩增子绝对定量测序项目文章再次登陆《Science of the Total Environment》

中国科学院南京土壤所王辉研究员课题组与南京农业大学生科院崔中利教授课题组合作的研究成果近期发表在环境科学与生态学TOP期刊《Science of the Total Environment》上,研究成果发现长期施氮降低了捕食性粘细菌的相对丰度和绝对拷贝数,改变了土壤中粘细菌群落结构。在这项研究中使用了天昊生物创新型的的微生物16S扩增子绝对定量测序技术。在恭喜客户发表文章同时,我们想跟大家分享一下文章的研究思路。
英文题目:Long-term nitrogen application decreases the abundance and copy number of predatory myxobacteria and alters the myxobacterial community structure in the soil
中文题目:长期施氮降低了捕食性粘细菌的相对丰度和绝对拷贝数,改变了土壤中粘细菌群落结构
期刊名:Science of the Total Environment
影响因子: 5.589
研究概要:
粘细菌是一种具有特殊社会生活方式(Social lifestyle)的微生物捕食者,这种社会性细菌(Social bacteria)在细菌域是独一无二的。这些捕食者在土壤微生物食物网中代谢活跃,具有驱动土壤微生物种群的能力。然而,施肥处理对农业系统中捕食性粘细菌的影响常常被忽视。本文采用高通量绝对丰度定量方法,对湖南祁阳红壤试验站施肥29年的农田中的粘细菌相对丰度和绝对拷贝数进行了研究。利用16S扩增子绝对定量测序技术,共检测到419个粘细菌OTUs,占细菌总丰度的0.25-2.70%。PCoA、ANOSIM和Manhattan analysis结果表明,施氮(N簇)和施猪粪(M簇)的粘细菌群落结构存在显著差异(p<0.05)。施加氮肥显著降低了粘细菌的相对丰度、拷贝数、物种积累指数和Shannon指数(p<0.05)。UpSet plots结果表明,氮肥处理的粘细菌OTUs数量仅为猪粪(M)处理的24.4%,施氮造成低丰度粘细菌OTUs数量的减少。此外,网络分析、RDA分析和随机森林RF分析表明,粘细菌相对丰度和绝对拷贝数是预测土壤细菌群落和功能基因α-和β-多样性的重要变量(P<0.05)。研究结果表明,施氮肥引起的土壤酸化是导致土壤粘细菌相对丰度和绝对拷贝数下降的最主要原因;粘细菌相对丰度和绝对拷贝数的变化与细菌的α-和β-多样性指数的变化显著相关,粘细菌群落的变化可能是影响整体微生物群落变化的重要因素。
研究背景:
捕食是影响生态系统群落结构和功能的重要因素之一。捕食性细菌与土壤微生物的群落结构和功能密切相关,粘细菌(myxococacales)是土壤生态系统中最常见(最丰富)的捕食性细菌。粘细菌可捕食土壤中的各类微生物,包括革兰氏阴性和革兰氏阳性细菌和真菌。粘细菌栖息于各种各样的环境中,如温带、热带雨林、北极苔原、沙漠、酸性土壤、海洋和其它盐碱环境,但其主要分布在土壤中。粘细菌是重要的土著捕食性细菌,以各种土壤微生物(死亡或存活)为食。粘细菌目前分为3个亚目(Cytobacterineae、Sorangiineae、Nannocystineae),10个科,29个属, 58个种。根据其不同的食性,粘细菌可分为两类:第一类是溶菌类群,其中包括绝大多数可培养粘细菌,这些粘细菌被称为微生物的微型掠食者,因为它们能够有效地降解其他细菌甚至真菌细胞(活细胞和死细胞)。第二类是降解纤维素类群,目前包括两个属,即Sorangium和Byssovorax,它们不能直接以活细菌为食,但可以降解死细菌。事实上,粘细菌多样性的研究已经取得了很大的进展。一些研究人员通过测定菌落和子实体的数量来估计粘细菌的细胞密度,结果表明,英格兰南部1g土壤中的溶菌粘细菌细胞数为2000~75000个,加拿大东南部1g土壤中的粘球菌和珊瑚球菌细胞数为1000~45000个。然而,这些研究通常只统计一个或几个粘细菌属,因此大大低估了粘细菌的数量。利用粘细菌特异性探针进行杂交分析的研究表明,粘细菌占细菌群落的比例不到1%。用粘细菌特异性引物W1/802R(用于亚目Cystobacteraceae, SDUCV34)和917F/W6RC(用于Sorangiineae亚目,SDUSV678)对土壤样品中粘细菌类群的丰度进行了评价,结果表明,在土壤生态位上,粘细菌群落具有高度多样性,几乎包括所有可培养的粘菌科或属。然而这些引物并不是所有粘细菌的通用引物,其覆盖率有待提高,特别是没有针对Nannocystineae的特异性引物。粘细菌在农业生态系统中的细胞密度、状态和功能需要进一步研究,我们对施肥系统如何影响土壤粘细菌种群缺乏全面的了解。我们认为利用通用引物对16S rRNA基因进行高通量测序仍是研究粘细菌群落结构最直接、最有效的方法:首先,通用引物可以覆盖广泛的粘细菌种类;第二,增加高通量测序的深度可以产生足够数量的粘细菌序列,同时提供关于每个粘细菌OTUs的丰度和拷贝数的信息;最后,16S rRNA高通量测序提供了一些功能预测信息。在本研究中,我们提出一种高通量绝对丰度测序(HAAQ)方法,利用人工合成的DNA内标(直接加入到土壤总DNA中)同时获得土壤细菌群落的绝对丰度和相对丰度。
粘细菌是重要的捕食性细菌,是农业土壤微生物食物链的重要参与者,加强对土壤生态中粘细菌群落的研究,对于了解粘细菌在农业生态系统中的地位和潜在作用具有指导意义。但是,目前关于长期施肥条件下农业土壤中粘细菌的相对丰度和绝对丰度的报道仍很少。本研究以中国农业科学院红壤试验站29年的试验田为研究对象,对粘细菌的多样性进行了研究,就以下几个方面提出了一系列问题:显著增加/减少土壤中粘细菌相对丰度和绝对拷贝数的因素(特别是与施肥有关的因素)是什么;粘细菌相对丰度和绝对拷贝数变化与土壤生态功能的关系是什么;粘细菌参与土壤微生物食物网的潜力及其与土壤微生物总体变化的显著相关性能否通过数据分析预测。
研究方法:
实验设计:
长期试验田于1990年在中国农业科学院祁阳红壤实验站建立,设计了12种常用施肥处理:1)不施肥对照(control, CK); 2)氮化肥 (N); 3)氮磷化肥 (NP); 4)氮钾化肥 (NK); 5)磷钾化肥 (PK);6)氮磷钾化肥 (NPK);7)氮磷钾化肥配施猪粪 (NPKM) ;8) 1.5倍用量的氮磷钾化肥配施猪粪(1.5NPKM);9)氮磷钾化肥配施秸秆;10)氮磷钾化肥配施猪粪+大豆轮作(NPKMR);11)猪粪(M);12)休耕(Nature)。每种处理共采集四个土壤样品(生物学重复),随机选择三个样本用于进一步研究。
16S扩增子绝对定量测序:
使用FastDNA® SPIN Kit for soil (MP Biomedicals, Santa Ana, CA)提取土壤DNA,然后在上海天昊生物科技有限公司进行16S扩增子绝对定量测序(V4-V5区域)。
研究结果:
基于测序结果,419个OTUs被注释为粘细菌,占所有细菌的0.25-2.70%。每g土壤中粘细菌细胞的平均数约为95994122(1,576,240-266,559,840)。每个土壤样品中检测到约277-3,825条粘细菌序列,平均2065个。每克土壤中的粘细菌OTUs平均拷贝数为229,063.8 (562.4-6,585,170)。419粘细菌OTUs,除No_rank和unassigned OTUs外,共分为3个亚目,10个科(100%已知科),29个属(51.7%)。只检测到4个属于降解纤维素类群的 OTUs:3个Sorangium OTUs和1个Byssovorax OTU。根据OTUs的数量,科排名如下:unassigned粘细菌(141个,占33.6%)、No_rank粘细菌(90个;21.5%)、Cystobacteraceae(41个;9.8%)、Polyangiaceae(40个;9.5%)、Labilitrichaceae (31;7.4%)、Sandaracinaceae (19; 4.5%)(表S2)。根据每克土壤的拷贝数,科排名如下:unassigned粘细菌(23,429,213;占24.4%)、No_rank粘细菌(19,668,760;20.5%)、Cystobacteraceae (14,258,960;14.9%)、Labilitrichaceae (10,429,414;10.9%)、 Polyangiaceae (9,026,838;9.4%)、Haliangiaceae (5,874,967;6.1%)。
施肥处理对土壤性质、土壤细菌群落α-多样性和粘细菌群落的影响
土壤理化性质测定结果表明,施用猪粪(M)的土壤pH、总氮、总磷、速效磷、速效钾和有机质含量均显著高于施用N肥的土壤,而总钾的情况则不同。经LEfSe分析,在这12种施肥处理下,共有1115个细菌类群存在显著差异(LDA>2,p<0.05),其中包括大量粘细菌和其潜在的猎物细菌。土壤微生物群落的ACE和Shannon指数因施用N肥(N、NP、NK、NPK和NPKS)而显著降低。相似的结果表明,施氮显著降低了粘细菌的相对丰度、拷贝数和细菌群落的总拷贝数。基于这些指标对猪粪(M)和N肥处理的一致响应,进行了一系列相关分析。结果表明,土壤pH、OM、TN、TP、AP、AK和土壤细菌群落α-多样性指数与粘细菌相对丰度和拷贝数呈显著正相关。
施肥处理对土壤粘细菌群落α-和β-多样性的影响
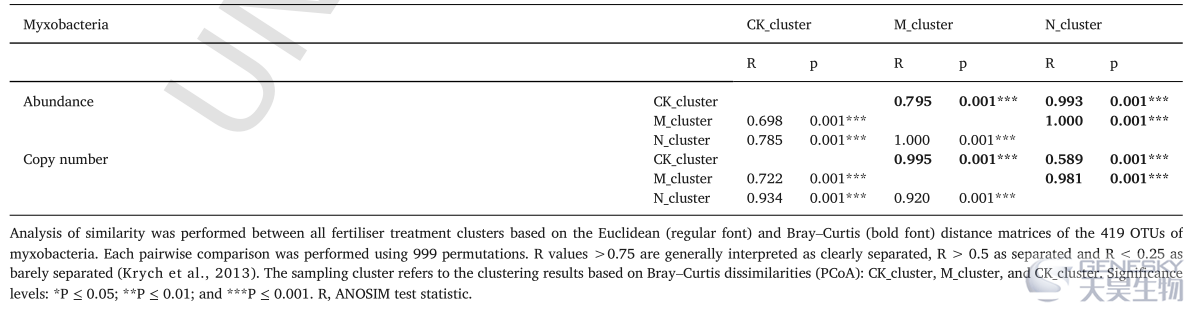
选取419个粘细菌OTUs进一步进行粘细菌群落的α-多样性、PCoA和ANOSIM分析,发现不同施肥处理间,尤其是N和M处理间,Shannon、Simpson和物种积累指数存在显著差异。粘细菌的群落用Bray-Curtis距离划分了三个主要簇(PCoA图A、B,N簇、M簇和CK簇)。PCoA和ANOSIM(表1)结果表明,M和N施肥处理对粘细菌群落(相对丰度和拷贝数)相似性距离具有有显著性影响,PK处理更类似于CK_簇样品(图1A,粘细菌相对丰度),而不是N_簇样品(图1B,粘细菌拷贝数),为了便于进一步分析,将PK分类到CK簇。此外,粘细菌相对丰度和拷贝数的PCoA轴1呈显著的线性相关关系(图1C,p<0.001),施肥处理对粘细菌相对丰度和拷贝数的群落结构影响相似。
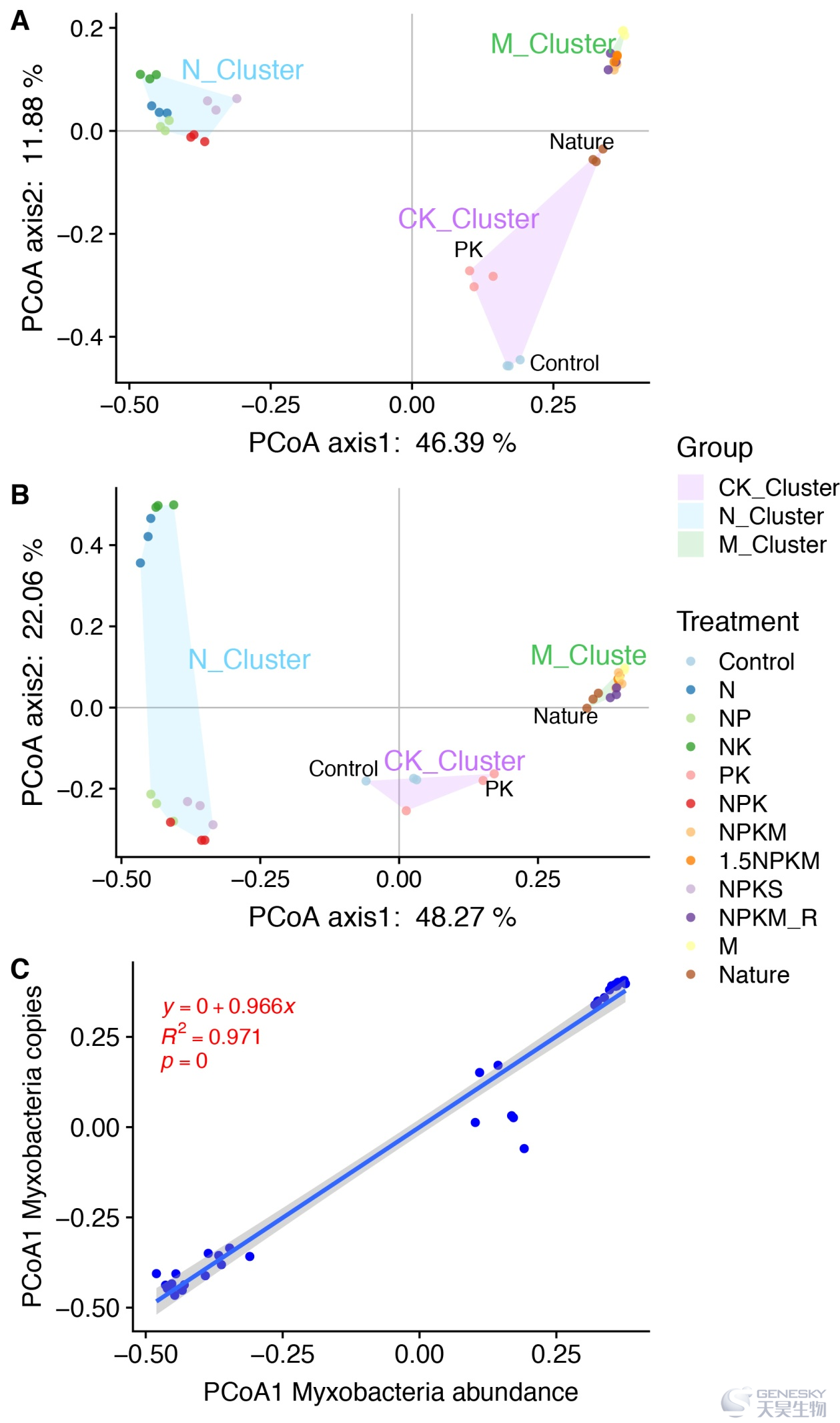
图1 施肥处理对土壤粘细菌群落相对丰度(A)和拷贝数(B)的影响,以及基于相对丰度与拷贝数PCoA 轴1的相关关系(C)。
表1 三种肥料处理组间的ANOSIM分析。
最高丰度的前100个粘细菌OTUs被定义为核心粘细菌OTUs,用于构建系统发育树(图2)。核心OTUs占粘细菌相对丰度的86.82%,占419个OTUs拷贝数的85.19%。前100位OTUs覆盖粘细菌的3个亚目和10个科,包括26个No_rank OTUs,19个unassigned OTUs,13个Polyan-giaceae OTUs,12个Labilitrichaceae OTUs和10个Cystobacteraceae OTUs等。几种优势OTUs的相对丰度和绝对拷贝数均高于大多数其它OTUs,但其它OTUs的丰度和拷贝数相对均匀,这些优势OTUs在分类学上也被分为不同的科或属,例如OTU154、OTU159、OTU1818、OTU285、OTU1469和OTU61 (图2)。
对所有419个粘细菌OTUs进行指示物种分析,物种指标分析显示,CK聚类样本中有46个指示OTUs,M聚类样本中有64个指示OTUs,N聚类样本中有9个指示OTUs。然而大多数指示OTUs不在前100(核心)OTUs中,图2中仅绘制了13个核心指示OTUs,分别是属于CK簇的OTU5432、OTU5651和OTU6121;属于M簇的OTU1987、OTU3197、OTU2670、OTU1527、OTU790、OTU3150、OTU1426和OTU1024;属于N簇的OTU9510和OTU10088。
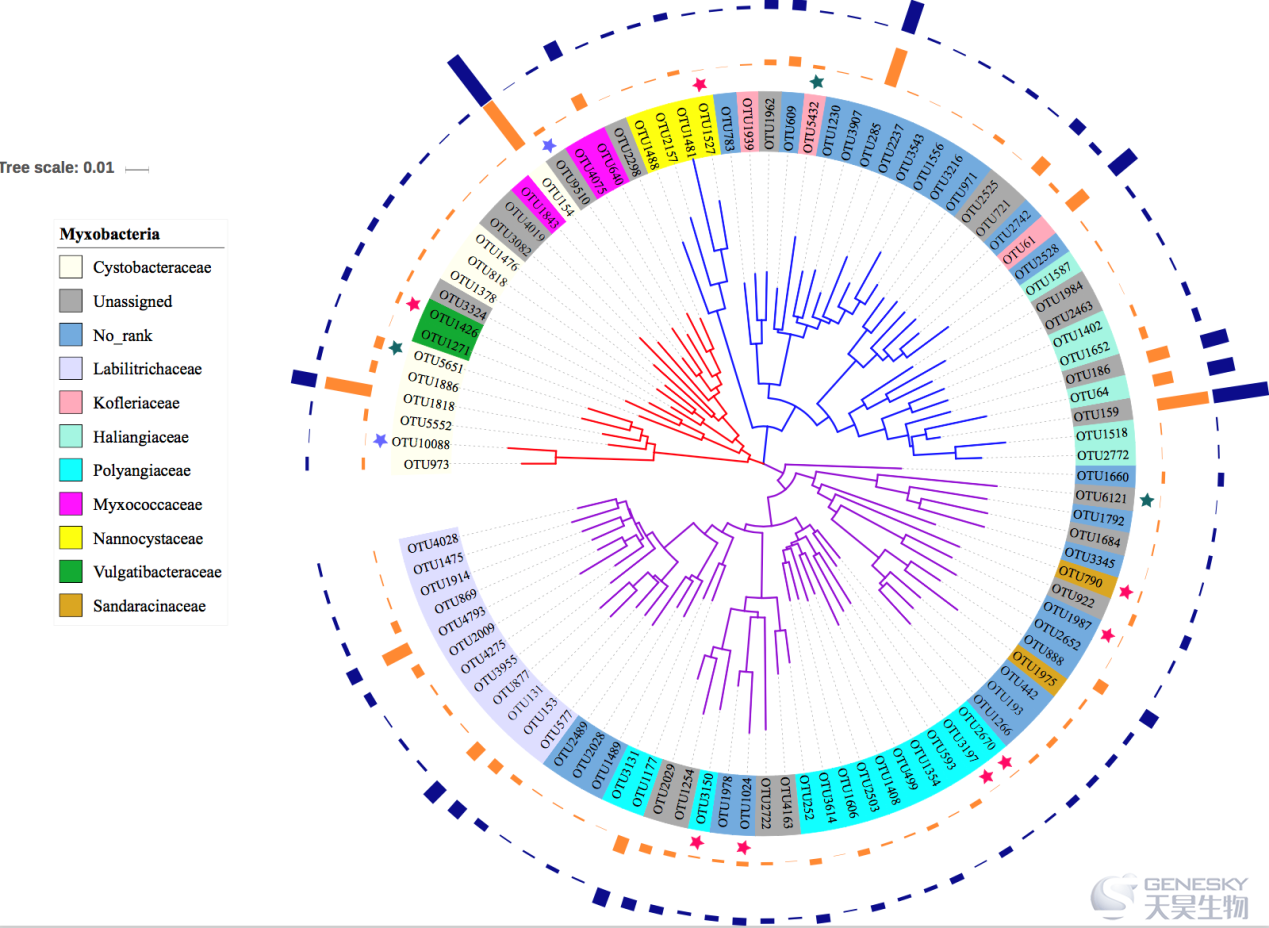
图2 粘细菌群落对CK、M、N簇土壤的响应。彩色分类树状图显示核心粘细菌(TOP100 OTUs),按亚目着色。内圈代表分类树(按科着色)。绿色星表示对CK簇土壤有显著响应的指示OTUs,红色星表示对M簇土壤有显著响应的指示OTUs,蓝色星表示对N簇土壤有显著响应的指示OTUs。
采用LEfSe对3个簇中相对丰度和拷贝数均存在显著差异的粘细菌进行评价,我们选择LDA得分高于2的细菌群进行鉴别,其差异具有统计学意义,结果发现相对丰度和拷贝数的LEfSe结果非常相似:相同的8个粘细菌科(除了No_rank)在不同的簇之间有显著差异,只有少数物种的结果不太一致。M簇中有9个主要的粘细菌属富集,分别是Kofleria, Myxococcus, Nannocystis, Sorangium, Enhygromyxa, Polyangium, Sandaracinus, Haliangium和No_rank粘细菌。在N簇土壤中富集的粘细菌属有Anaeromyxobacter和 Vulgatibacter,CK簇土壤中富集的是Polyangium, Sorangium, Byssovorax, Phaselicystis。
粘细菌OTUs在M和N簇中的差异
通过按照分类法(科/属)排列419个OTUs,并在曼哈顿图中显示N簇和M簇的相对丰度(图3A)或绝对拷贝数(图3B),来剖析观察到的粘细菌群落变化,结果显示,在N和M处理之间,粘细菌OTUs相对丰度或绝对拷贝数有显著差异(图3)。M组有189个OTUs(相对丰度)和229个OTUs(绝对拷贝数)富集,而N组只有48个(相对丰度)和52个OTUs(绝对拷贝数)富集,这些显著富集的OTUs的相对丰度和绝对拷贝数高于大多数没有显著变化的OTUs(图3A)。结果表明,施加M肥显著增加了各科的优势粘细菌的相对丰度和绝对拷贝数,而一些稀有的粘细菌OTUs对M和N处理没有明显的响应。
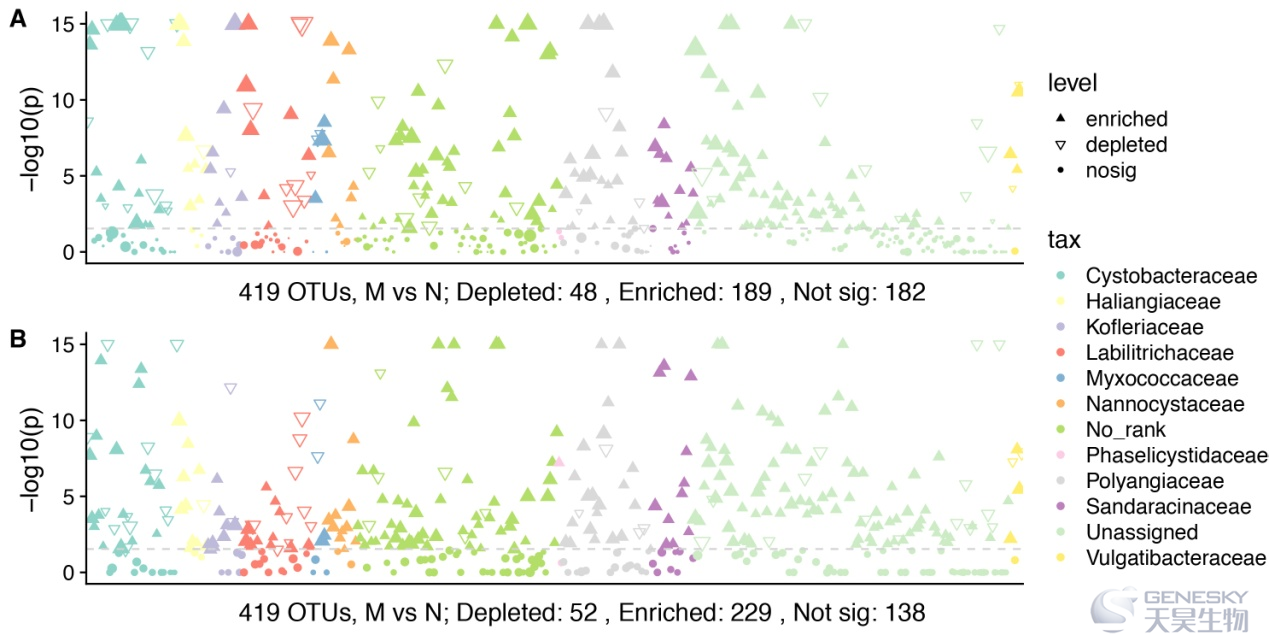
图3 曼哈顿图显示了M处理土壤相对于N处理土壤中富集的粘细菌的相对丰度(A)和绝对拷贝数(B)。显著富集的OTUs被描绘为填充三角形,显著减少的OTUs被描绘为空心三角形,不显著的OTUs被描绘为全圆。虚线对应于p值=0.05显著性阈值。每个点的颜色代表了OTU(科水平)的分类隶属关系,每个点的大小对应于36个土壤样品中OTU的平均相对丰度或绝对拷贝数。
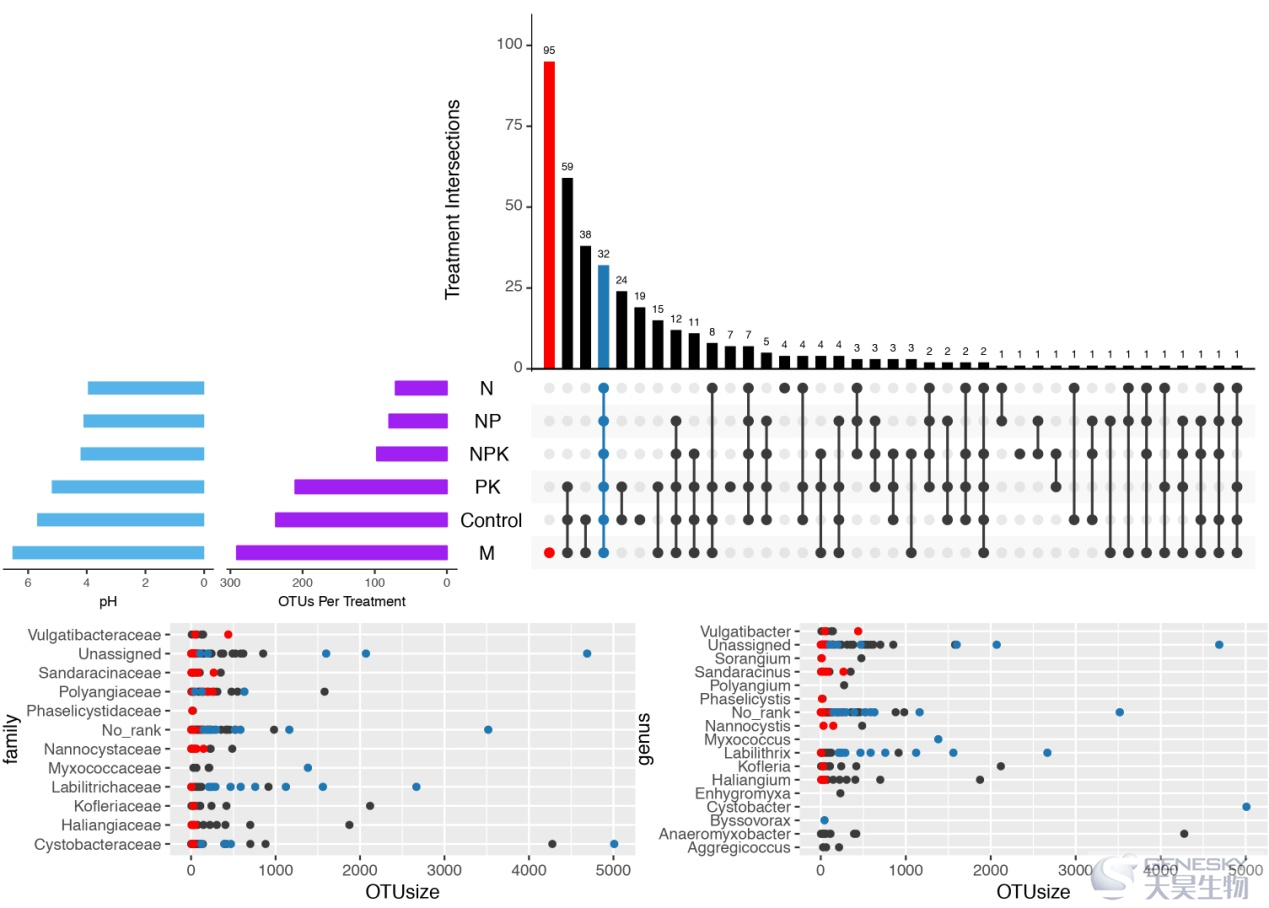
LEfSe和Manhattan(图3)的分析结果表明,施用M肥显著增加粘细菌OTUs的相对丰度或绝对拷贝数,而施用N肥则相反。选择UpSet plots分析进一步观察施N肥或施M肥对粘细菌OTU丰度和群落的影响,为了简化模型并减少变量数量,只选择CK、N、NP、PK、NPK和M施肥处理用于UpSet plots分析。结果表明,M肥处理的粘细菌OTUs数量最高(291个,占419个OTU总数的69.5%),其次是CK(237个,56.6%)、PK(210个,50.1%)、NPK(97个,23.2%)、NP(80个,19.1%)和N(71个,16.9%)处理(图4), 施N处理粘细菌OTUs仅为M处理的24.4%。此外,M肥处理独有的粘细菌OTUs数也最高(95;22.7%),其次是CK(19;4.5%)、PK(7;1.67%)、N(4)、NPK(1)和NP(0)处理。
散点图显示了这些样品中所有粘细菌OTUs的OTU size(丰度信息)和分类学信息(科和属)(图4)。M肥处理所特有的95个OTUs丰度较低,说明大多数是罕见(稀有)的OTUs,然而6组共有OTUs的丰度高于M肥处理所特有的OTUs,这些共有OTUs中核心OTUs数目也多于M肥处理所特有的OTUs,结果表明施M肥增加了稀有粘细菌OTUs的数量,而施N肥则减少了稀有OTUs的数量。
图4 UpSet结果显示不同处理下粘细菌OTUs的关系。左面(紫色条)显示每个处理粘细菌 OTUs的数量。矩阵中的黑圈表示交叉的处理集。M的单向交集(红色)和所有6个处理的六向交集(蓝色)。OTU丰度(OTUsize):该OTU在所有样品中的序列总数,可用于指示土壤样品中每个OTU的平均丰度。
土壤性质与粘细菌群落及功能的关系
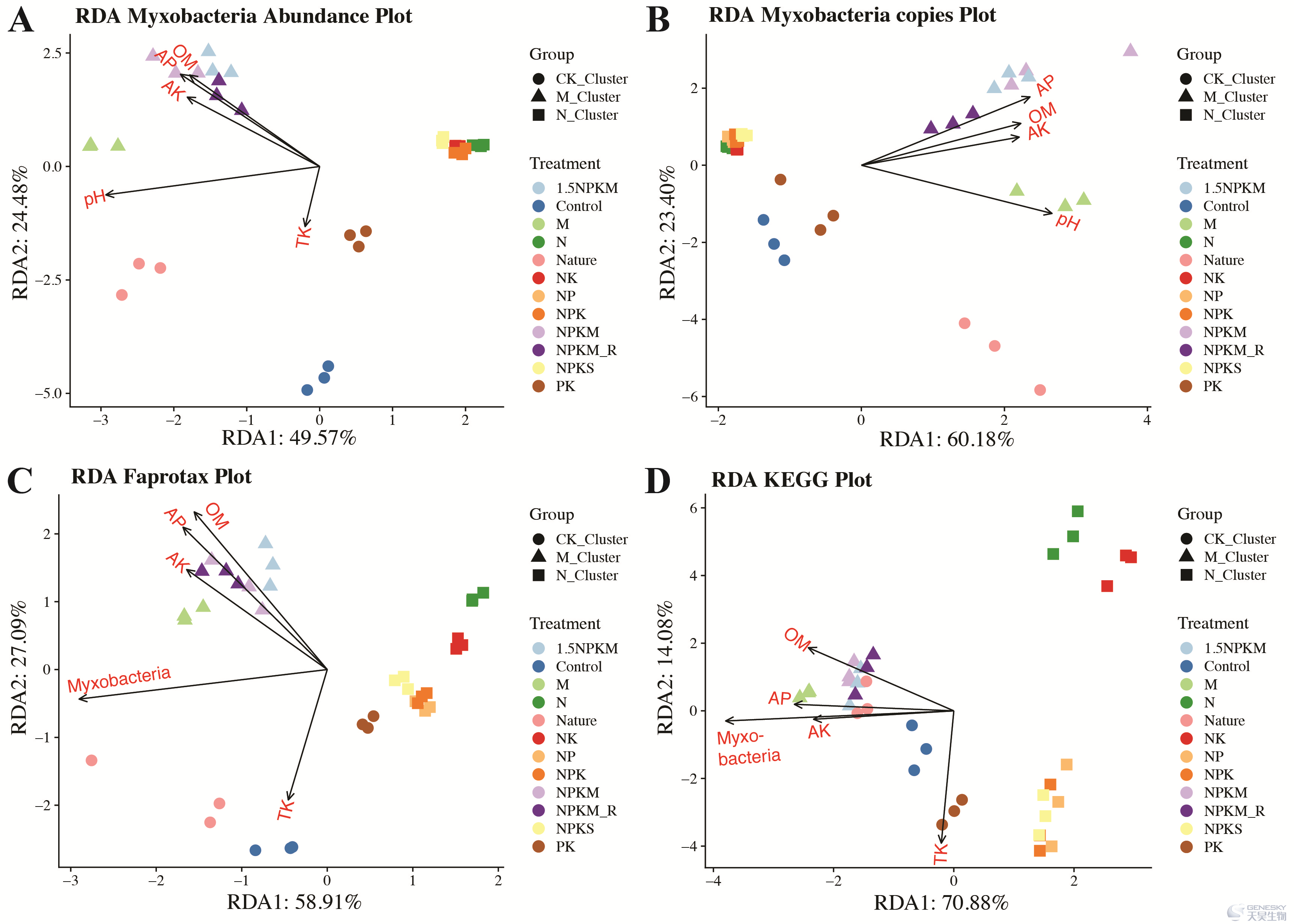
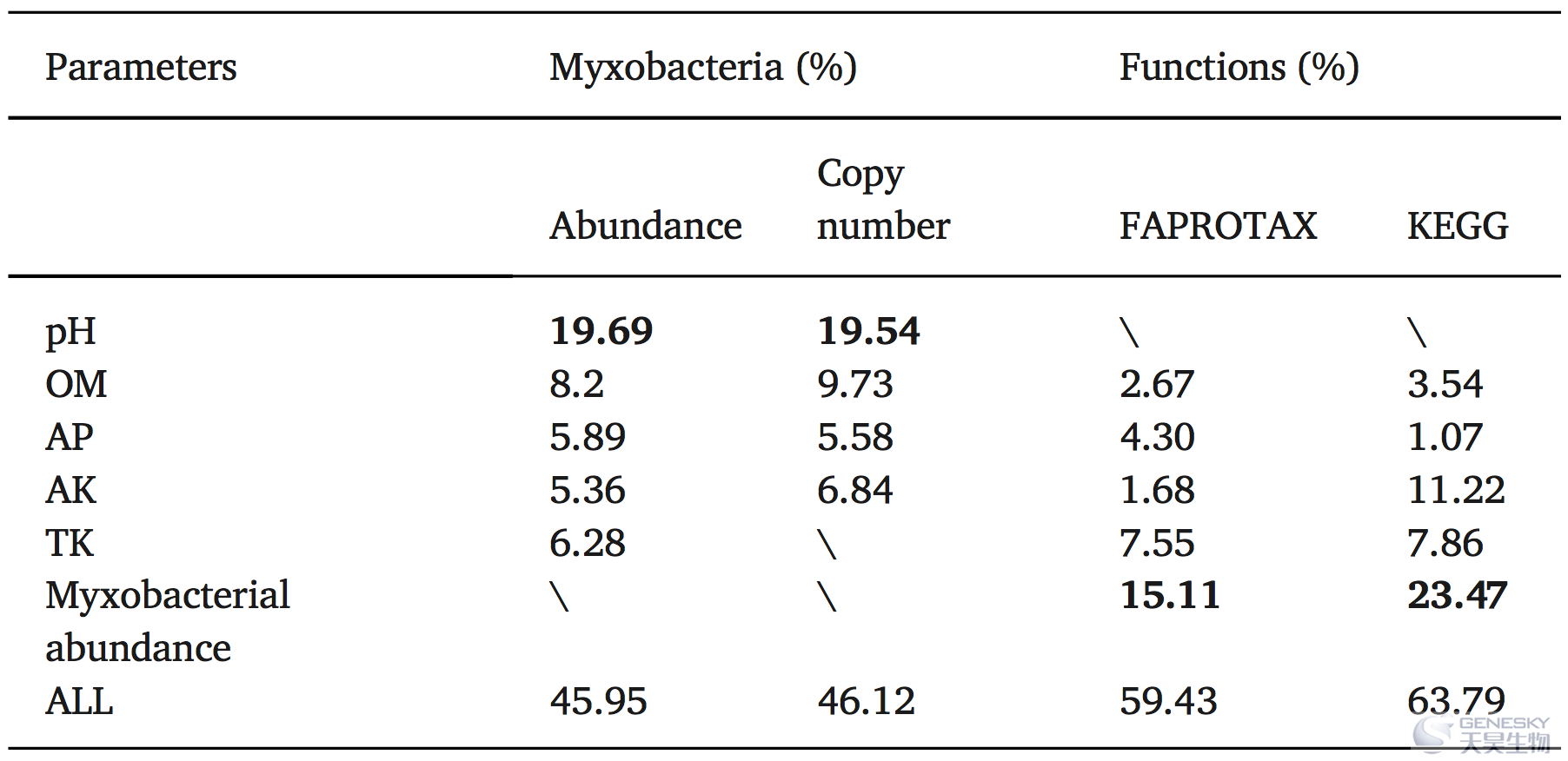
通过RDA分析以了解OTUs水平的粘细菌群落(图5A, B)和土壤功能结构(图5C, D)。依据蒙特卡罗检验和方差膨胀因子共筛选出4-5个具有显著性影响的参数,即pH、OM、TK、AP、AK、粘细菌丰度(图5)。粘细菌群落结构的RDA结果显示N簇样品可以与其它处理样品区分开来,pH值解释了最大的变异,解释了19.69%(图5A;相对丰度)和19.54%(图5B;绝对拷贝数),而OM、AP、AK和TK含量解释的变异逐渐减少(见表2)。在土壤功能结构方面,粘细菌丰度解释了土壤功能的最大变化,解释了15.11%(表2;FAPROTAX)和23.47%(表2;KEGG)的变化。
图5 基于粘细菌丰度(A)、粘细菌拷贝数(B)、FAPROTAX(C)和KEGG(D)的土壤样品之间的RDA分析。圆圈表示CK聚类;三角形表示M聚类;正方形表示N聚类;样本点的颜色表示12个施肥处理。
表2土壤参数和粘细菌丰度对粘细菌群落和土壤功能变异的贡献
土壤粘细菌群落及KEGG功能的分子生态网络分析
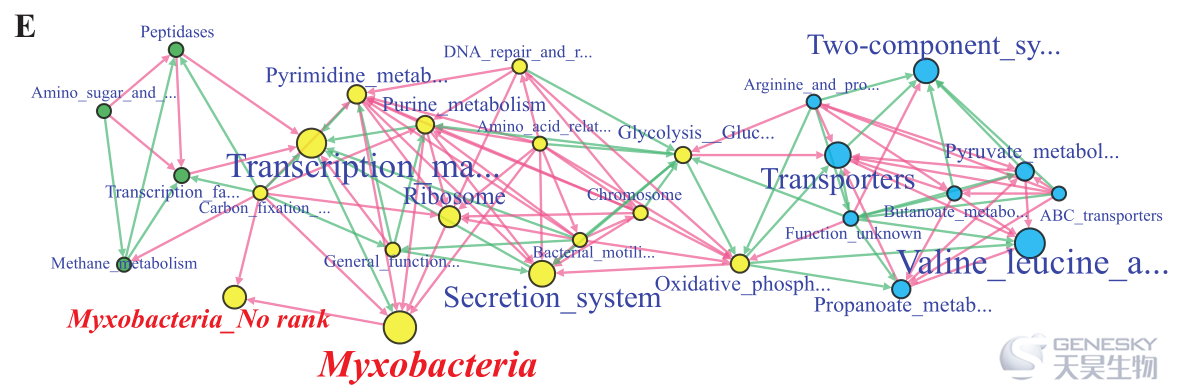
分子生态网络分析已被广泛应用于研究土壤微生物与功能基因的相互作用。利用粘细菌(包括29个粘细菌属)的丰度和KEGG功能建立共现性网络,结果表明,在生成的网络中只有粘细菌和粘细菌_No_rank的丰度与26个KEGG功能 共计28个节点,但在网络中未发现其它粘细菌属。粘细菌丰度在所有节点中具有最高的特征向量中心性(节点最大,图5E),此外粘细菌丰度节点的其它中心度指数都相对较高(Table S7)。KEGG节点与粘细菌节点存在共现性(co-occurred),这些KEGG功能参与重要的能量代谢过程,如氨基酸相关酶、原核生物的碳固定途径、嘌呤代谢、嘧啶代谢、核糖体和转录等(图5E)。
图5 粘细菌丰度与土壤功能的共现性网络分析(E,KEGG)。红色节点标签表示粘细菌,蓝色的节点标签表示KEGG代谢途径,模块随机着色。红色连接表示两个单独节点之间的显著正相关,绿色连接表示负相关。
土壤细菌群落的潜在驱动力及其功能
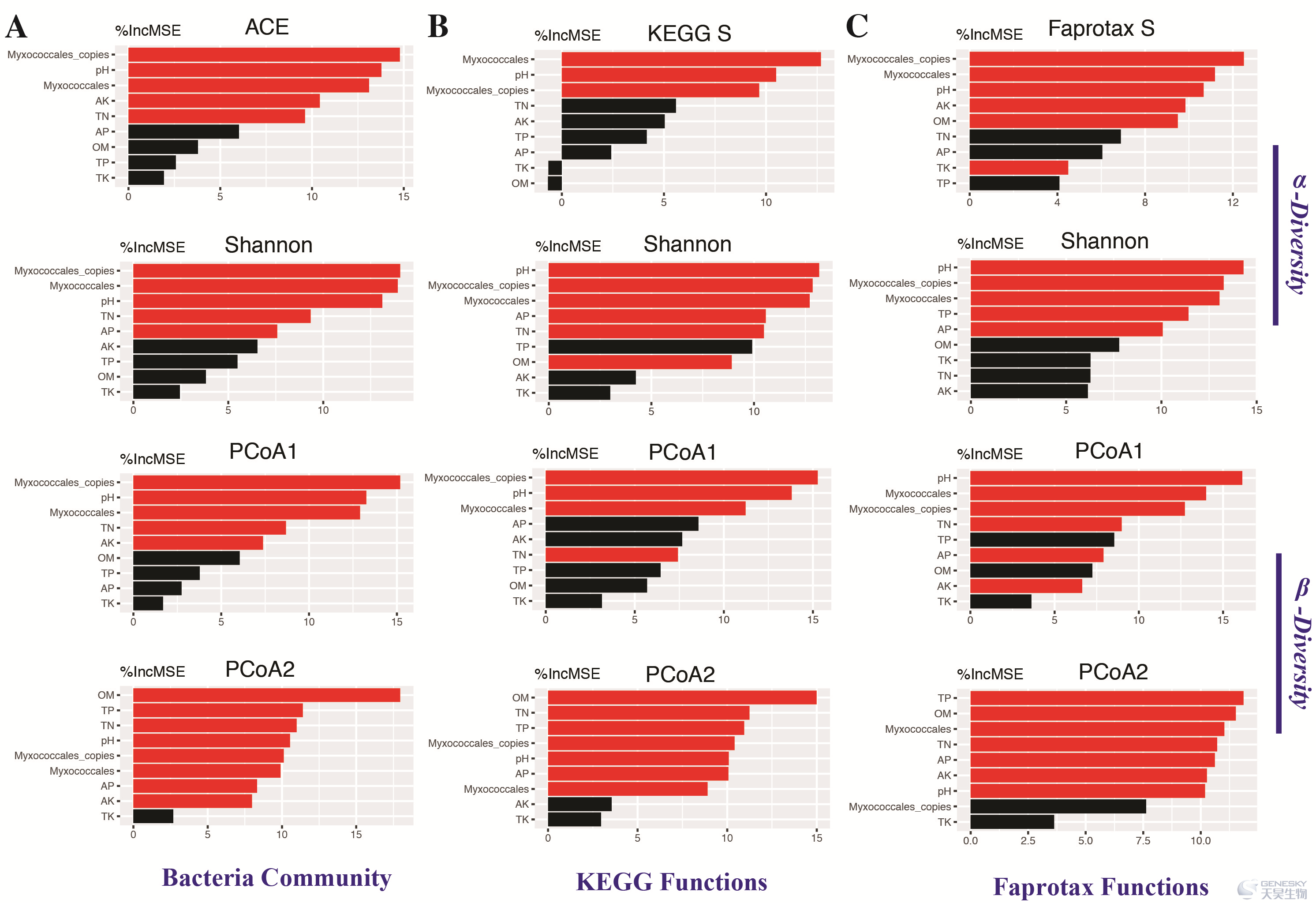
RF分析确定了土壤细菌群落α-和β-多样性及其功能指标的主要微生物预测因子(P<0.05;图6)。粘细菌的丰度、拷贝数和pH值是解释三种细菌群落α-和β-多样性及其功能的最重要变量(图6A、B、C)。这三个变量的MSE%值高于其它变量,相对较高的MSE%值表示相对更重要的变量。此外,TN是预测细菌群落和KEGG功能的重要变量(图6A、B)。TN也是预测PCoA1的重要变量(P<0.05)。AP是预测Shannon指数的重要变量(P<0.05)。
图6土壤理化对细菌群落(A),KEGG(B)和FAPROTAX(C)功能多样性的贡献。均方误差值增加的百分比(MSE%)意味着这些预测值的重要性。红色条表示显著预测(p<0.05),黑色条表示不显著预测。
常规扩增子测序技术和qPCR技术技术痛点
常规16S扩增子测序技术虽然其具有高通量,低花费,可客观还原菌群结构及相对丰度比例的巨大优势,但是其是通过某一OTU分类单元的序列数所占总序列数的比值来获得某个细菌的相对丰度比例信息,然而相对丰度信息不能反映样本中物种真实的绝对丰度情况,例如微生物某一类群的相对丰度比例增加不一定真正是相应微生物类群的绝对丰度增加,可能是其它微生物类群的绝对丰度减少是导致其在群落结构中相对丰度比例的增长,因此常规16S扩增子测序技术基于相对定量分析的错误结果解释可能导致假定的因果关系!
qPCR绝对定量技术虽然可以进行物种绝对定量分析,但是qPCR定量实验结果常常不稳定,且特定物种qPCR需要设计特定引物,对引物特异性要求较高,且引物优化难度较大,导致常规的qPCR绝对定量实验不再适用于组成较为复杂的环境样本微生物绝对定量。此外,当环境样本中往往含有腐殖酸,这些腐殖酸会通过抑制酶的活性抑制PCR,从而影响qPCR对细菌拷贝数定量结果的准确性。
天昊微生物16S扩增子绝对定量测序技术简介
该技术是一种将qPCR绝对定量技术和常规16S扩增子测序技术合二为一的技术,该技术不但可以进行Alpha多样性分析、群落组成分析、Beta多样性分析、指标和微生物相关性分析等常规16S扩增子测序分析,关键可以解析样本中总细菌的绝对拷贝数,还可以解析样本中每个物种的绝对拷贝数,因而对微生态学内许多悬而未决的问题具有进一步阐明的潜力。此外,该技术进行细菌拷贝数定量时,构建标准曲线的内标和样本DNA是在同一个样本孔中一起进行PCR反应,所以PCR反应效率相同,因此校正了腐殖酸对PCR的影响,避免了腐殖酸等PCR抑制物对样品细菌16S拷贝数定量的影响,因此针对土壤、水体和淤泥等环境样本,天昊生物16S扩增子绝对定量测序技术计算得到的细菌16S拷贝数相对于qPCR更准确。
天昊微生物16S扩增子绝对定量测序技术应用情况
目前天昊微生物16S扩增子绝对定量测序技术平台已经完成项目百余个,合作单位包括中国科学院微生物研究所、中国科学院南京土壤研究所、中国科学院水生生物研究所、同济大学环境科学与工程学院、厦门大学环境与生态学院、中国农业大学、南京农业大学、东北农业大学、重庆市农业科学院、盐城工学院、南京财经大学、南京中医药大学、武汉大学中南医院、新疆医科大学公共卫生学院、山东大学齐鲁医院等多个单位,覆盖环境土壤微生物,环境水体微生物和医学肠道微生物等多个领域,利用该技术的项目文章成功陆续发表在环境科学与生态学TOP期刊《Science of the Total Environment》(IF= 5.589)和应用化学1区期刊《Carbohydrate Polymers》(IF=6.044)上,目前该技术因其创新性、准确性和稳定性受到客户的广泛好评! 天昊生物目前是国内唯一一家提供 “微生物16S扩增子绝对定量测序”技术的服务商,热烈欢迎各位老师与我们交流沟通!
 咨询热线:400-065-6886
咨询热线:400-065-6886