 咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
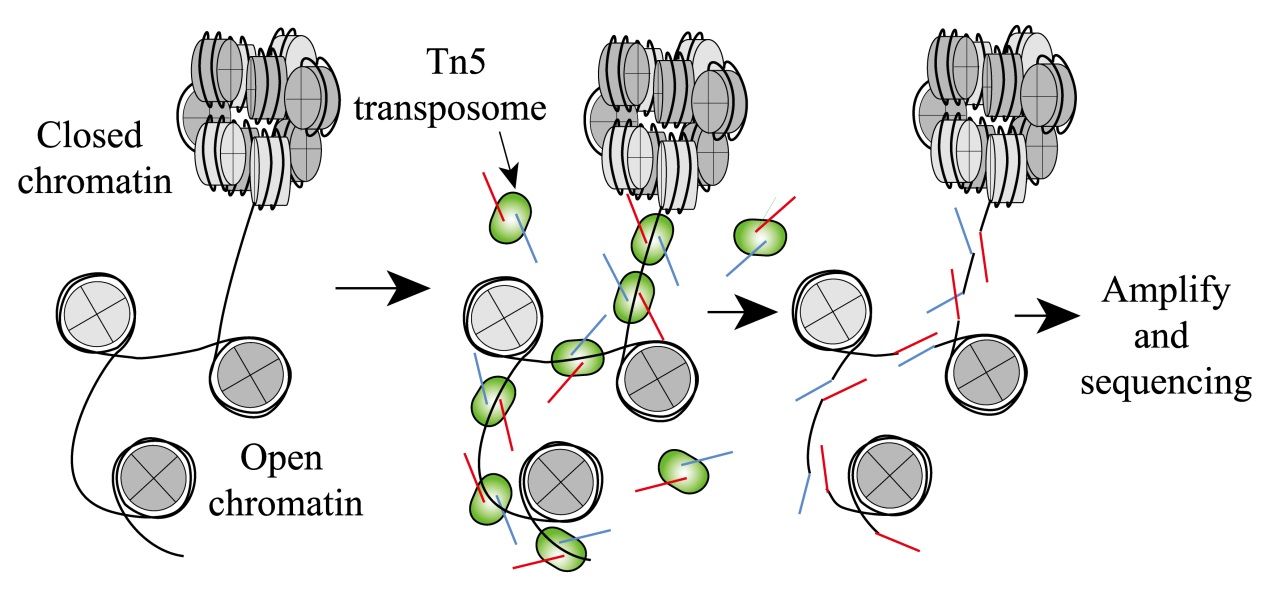
真核生物的染色质通常情况下以核小体为单位进行致密的折叠,不表现出转录活性,同时部分开放的染色质区域作为特异性反式作用因子(如转录因子,酶等)和顺式作用元件(如Enhancer,Insulator等)与基因组DNA相互作用的活跃区域。这种由染色质的开放程度定义的染色质可接近性(Chromatin Accessibility)对细胞的基因的表达,DNA的复制和修复等生命活动产生重要的影响,并且时刻受到细胞的严格调控。因此检测特定时空状态下细胞染色质的开放程度(从关闭状态到半开放,从半开放到完全开放),是探索染色质结构重塑(Chromatin Remodeling)对生物的生长发育、疾病的发生发展影响的重要研究方向。
ATAC-seq(Assay for Transposase-Accessible Chromatin using sequencing)方法利用高灵敏度的转座酶(Transposase,Tn5)在寻找染色质上可接近位置的同时对染色质DNA进行片段化和接头连接,扩增后进行高通量测序。细胞需求量在50000个左右,更适合数量有限的临床珍贵样本,实验操作上更方便快捷,是一种创新的单碱基分辨率的表观遗传学研究技术。目前该技术已被广泛应用于揭示染色质丰富的表观调控信息、不同状态下开放染色质的变化,以及多种疾病中开放染色质图谱的绘制等,同样适用于真核细胞重编程及单细胞染色质开放性的研究,在医学领域是重大疾病发病机制、药物作用机制、新药研发和生物标志物等研究的重要利器。

2019年6月来自华西医院的科研团队于Cancer Research杂志发表了题为“The Open Chromatin Landscape of Non-small Cell Lung Carcinoma”的文章,结合50例原发性非小细胞肺癌(NSCLC)患者的开放染色质数据(ATAC-seq)、全基因组测序数据(WGS)和RNA测序数据(RNA-seq)揭示了NSCLC的开放染色质图谱(Figure1A)。其中发现患者间开放染色质的异质性,异质性的程度与一些临床参数相关,而且肺腺癌(LUAD)和肺鳞癌(LUSC)间也显示出明显不同的开放染色质模式。
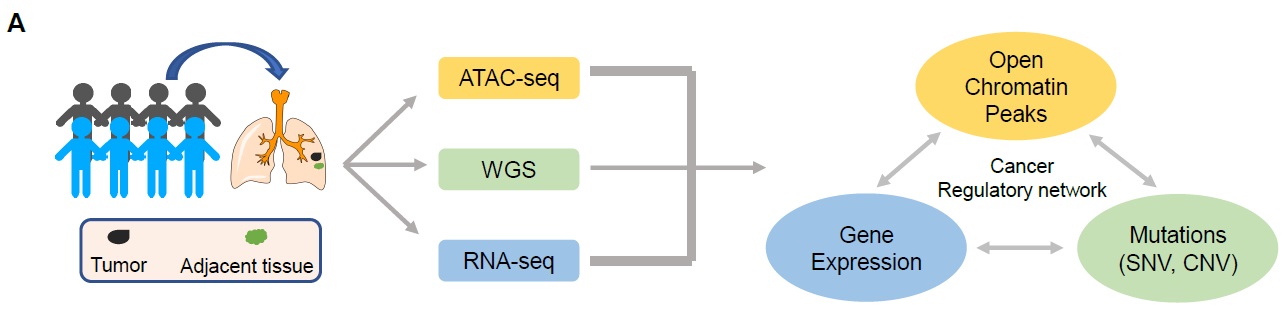
Figure1A 实验总体设计思路
1. 实验总体设计及样本突变特征
研究人员共收集了34例腺癌患者、13例鳞癌患者及4例非恶性结节患者(BSPN)的原发肿瘤样本,同时收集40例NSCLC及2例BSPN患者对应的癌旁样本供somatic突变calling(Figure1A)。其中绝大多数somatic SNVs和indels位于基因间区和内含子区(Figure1B)。与先前报道相似在NSCLC驱动基因中发现了一些somatic SNVs,包括TP53在26.92%的LUAD患者和33.33%的LUSC患者中,EGFR在19.23%的LUAD中,CSMD3在33.33%的LUSC中,NFE2L2在33.33%的LUSC中(Figure1C)。所有的LUSC样本与富集的突变特征S3(与COSMIC signature 5类同)相关,而大多数LUAD样本与突变特征S2(与COSMIC signature 9类同)相关(Figure1D)。基于四种突变特征的贡献度将所有样本进行聚类,绝大多数样本聚类结果较好(Figure1E)。同时在这些样本中发现很多已知基因存在显著的扩增或缺失(Figure1F)。
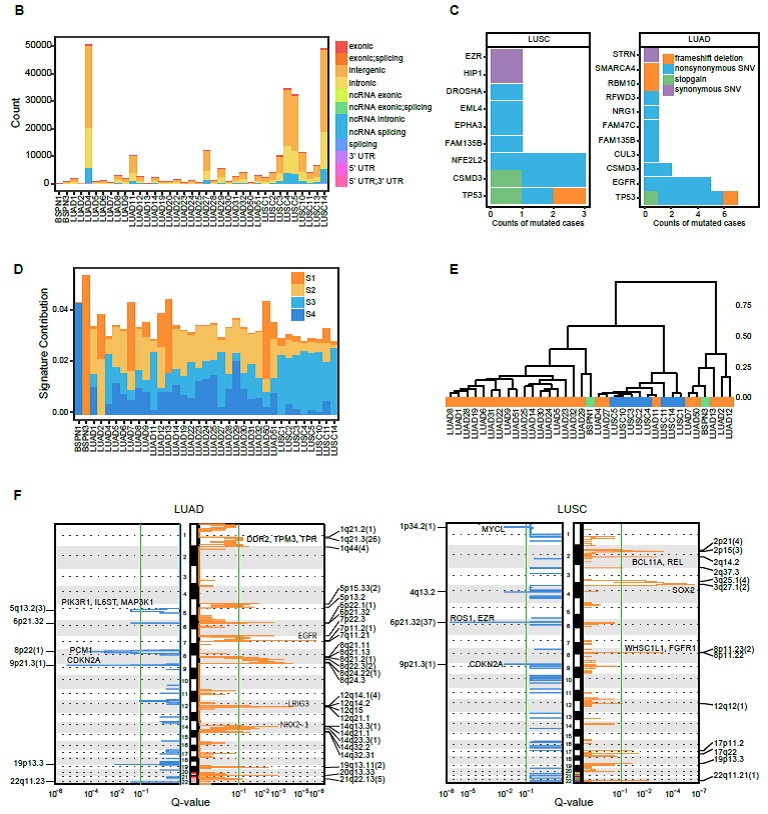
Figure1B-F NSCLC患者突变谱
2. NSCLC开放染色质特征
为了揭示开放染色质区域,利用ATAC-seq对每个肿瘤样本进行测序。采用Jaccard index score评价样本间开放染色质水平的异质性,发现NSCLC样本间异质性比干细胞或分选的血液细胞显著更高(Figure2A)。为了揭示样本特定的peak能否显示功能的特异性,利用少数peaks(minority peaks,在少于20%的样本中出现)对样本进行分类。有意思的是少数peaks与病理类型显著相关,LUSC样本包含更多的minority peaks,另外minority peaks的比例与更高的肿瘤分期、吸烟史、女性显著相关,但与peak数、转移与否和年龄无显著相关性(Figure2B)。
为了探索整体的开放染色质分布作为markers区分NSCLC样本,利用ATAC-seq信号将这些样本聚类成3个cluster(Figure2C)。其中cluster1有4个LUSC和4个LUAD样本组成,这些样本有显著更少的开放染色质peaks以及更高的样本间异质性(Figure2D),同时显示出患者特异性的特征,而不是癌症类型特征。Cluster2主要由LUSC样本组成,开放染色质的TSS上下游2.5kb范围发现了310个基因具有上调的表达,这些基因主要富集在角质化过程(Figure2E),这也是LUSC特定的现象,而非LUAD。Cluster3主要由LUAD样本组成,可以进一步分为3个sub-cluster,这三个sub-cluster有明显不同的开放染色质模式和不同的肿瘤分期(Figure2C)。
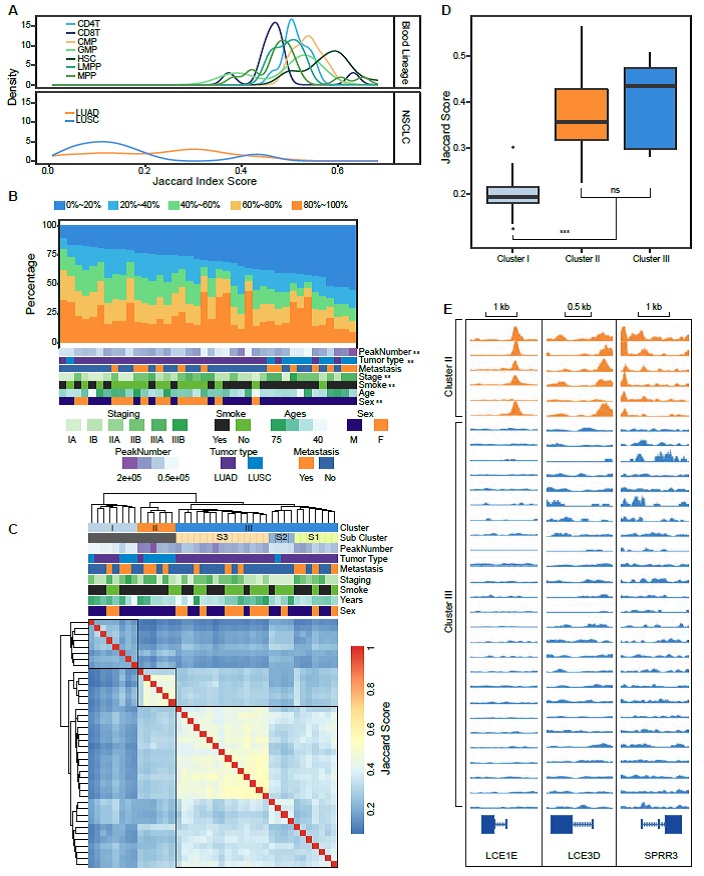
Figure2 NSCLC开放染色质图谱
为了更好地理解瘤内异质性,对一例LUSC患者进行了单细胞ATAC-seq,对质控后的1651个细胞聚类发现7个主要的cluster,其中clutser3的开放染色质特征与bulk样本具有高度一致性,97%的peaks出现在了bulk样本中。因此在单细胞水平上观察到一些开放染色质区域的高度异质性(Figure3)。
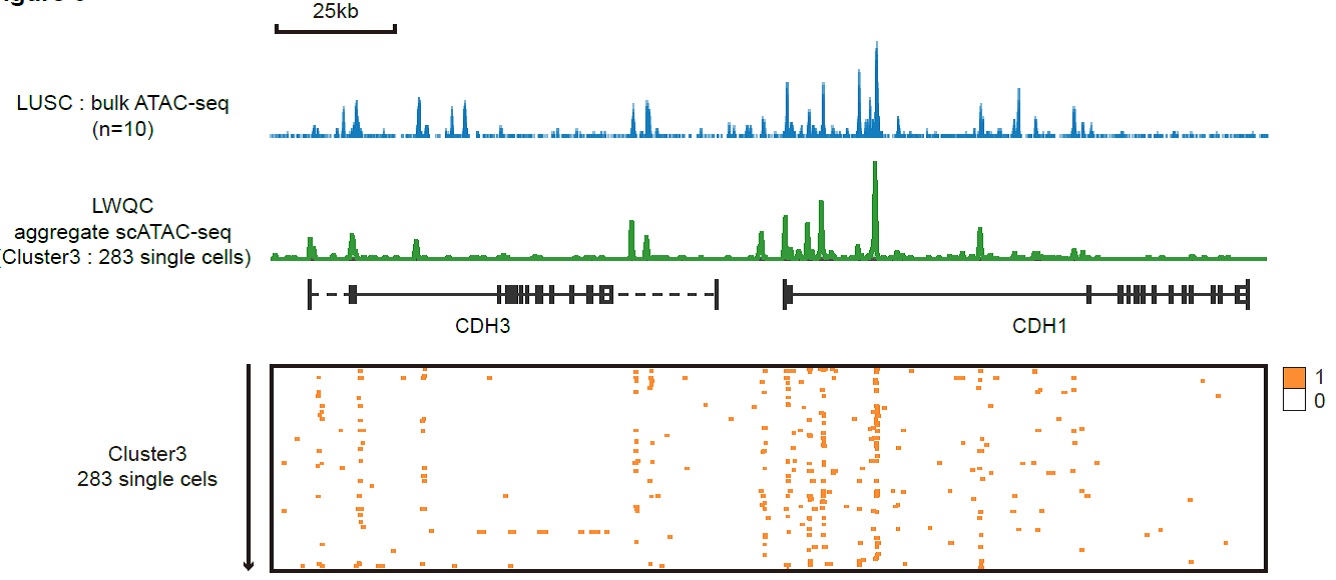
Figure3 肿瘤单细胞ATAC-seq展示
3. 宽的开放染色质peaks与NSCLC关键基因相关
整合了白血病的ATAC-seq数据进行分析,发现宽的开放染色质peak相关的基因中,310个是NSCLC特异的,368个是Leukemia特异的,337个是共有的(Figure4A)。其中在3个样本中EGFR的几乎整个gene body区被开放染色质区覆盖(Figure4B)。另外,宽的开放染色质peak相关的基因平均表达水平高于其它基因,但是仅38个基因具有显著的差异表达(Figure4C)。宽的开放染色质peak相关的基因其表达水平变异系数更高(Figure4D)。
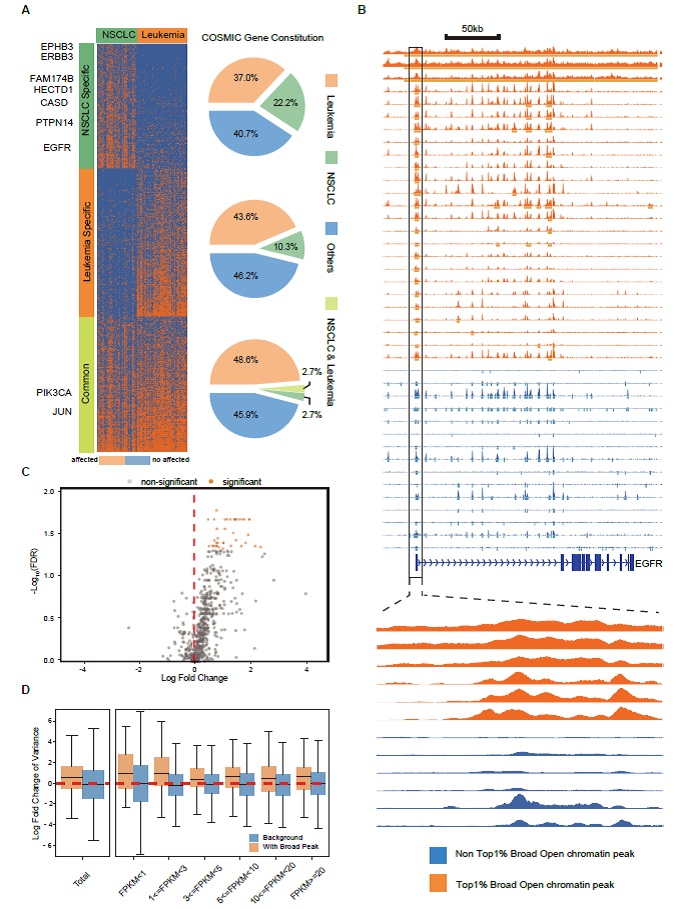
Figure4 NSCLC宽的开放染色质区域
4. Somatic CNV/germline SNV与开放染色质及基因表达间的关系
文章最后整合多组学数据从somatic CNV的角度分析与开放染色质关系,同时结合基因表达数据发现同时具有somatic CNV和开放染色质的片段与基因表达有显著的相关性,CNV gain其表达水平显著更高,CNV loss其表达水平显著更低(详见原文)。
另外从QTL的角度整合germline SNV、ATAC数据和基因表达数据发现某些基因的信息,同时与GWAS数据整合分析某些风险位点(详见原文)。
总结讨论
文章通过整合NSCLC肿瘤组织的多组学数据显示这种类型的数据能够揭示一些基因调控网络,但是样本量的限制使得分析具有一定的局限性!
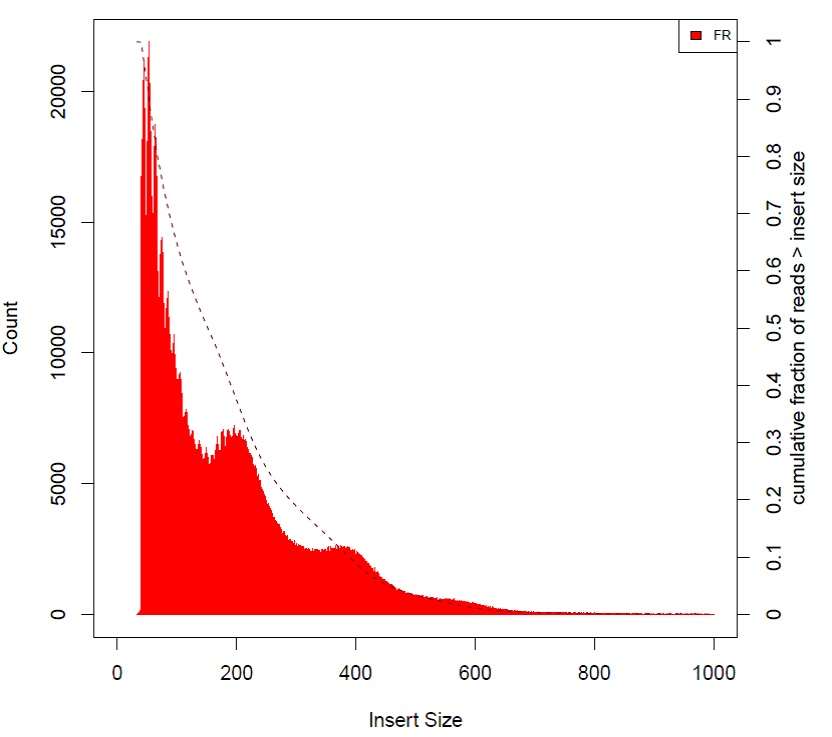
ATAC-seq片段大小揭示核小体片段信息
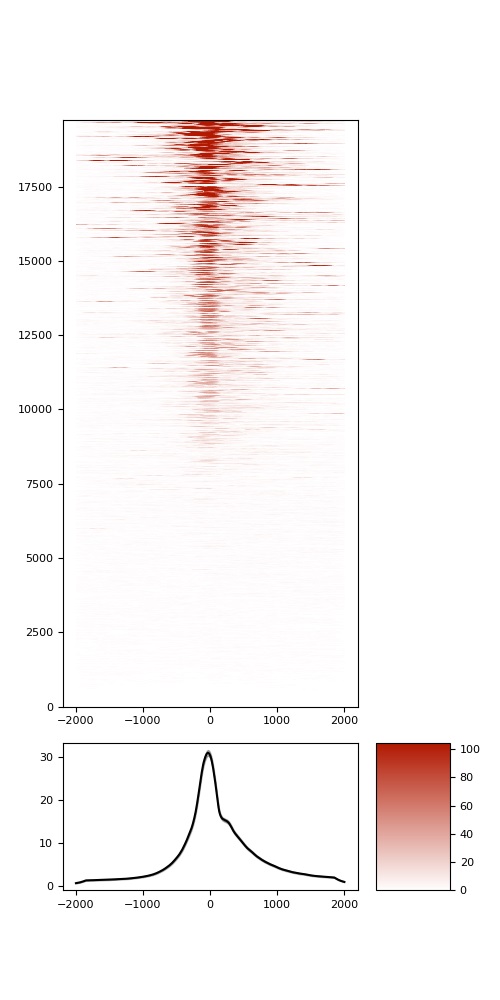
转录起始位点为中心的上下游2kb区域信号分布
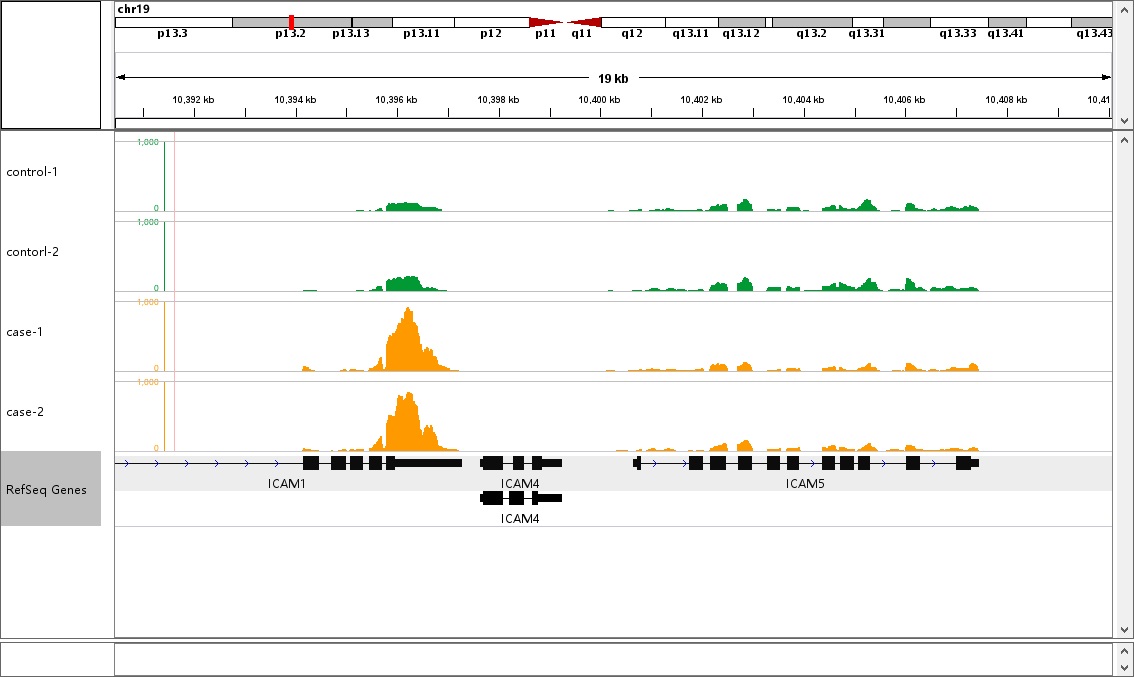
样本间差异peak可视化信息

