 咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
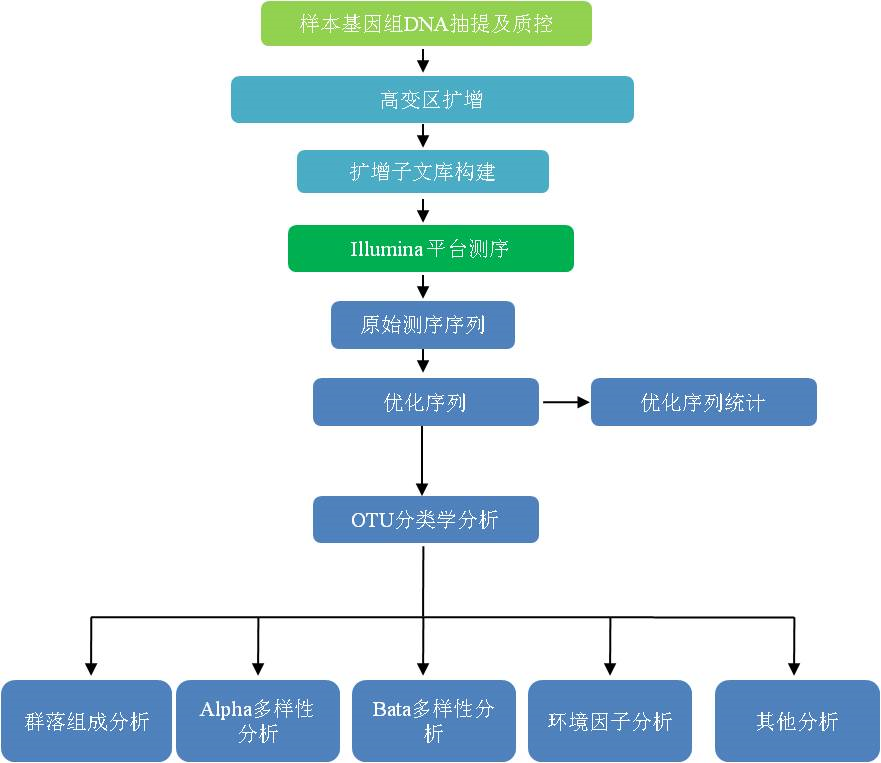
32只雄性C57BL/6小鼠分为4组(每组8只):第一组小鼠给予正常饮食喂养+200微升水(ND组)、第二组小鼠给予高脂饮食喂养+200微升水(HFD组)、第三组小鼠给予高脂饮食喂养+200微升的400 mg/kg/day苦丁茶(KDC)提取物(HFD-KDC组)、第四组小鼠给予高脂饮食喂养+200微升的400 mg/kg/day茯砖茶(FBT)提取物(HFD-FBT组)。每组小鼠驯化一周后灌胃8天。每周测量食物摄入量和体重。粪便、肝、血、肾周脂肪组织和附睾脂肪组织实验结束时收集,立即在液氮中冷冻保存在-80℃。
使用qPCR测定肝脏脂质代谢相关基因表达量:肉毒碱棕榈酰转移酶-1(CPT-1)、过氧化物酶体增生物激活受体α(PPARα)、过氧化物酶体增生物激活受体γ(PPARγ)、固醇调节元件结合蛋白-1c(SREBP-1c)、脂肪酸合酶(FAS)、肝X受体α(LXRα)。
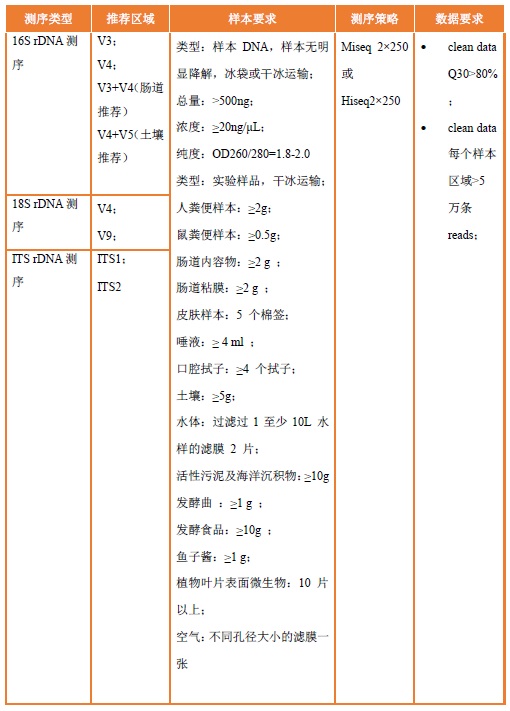
细菌16S扩增子测序区段为:(V4);
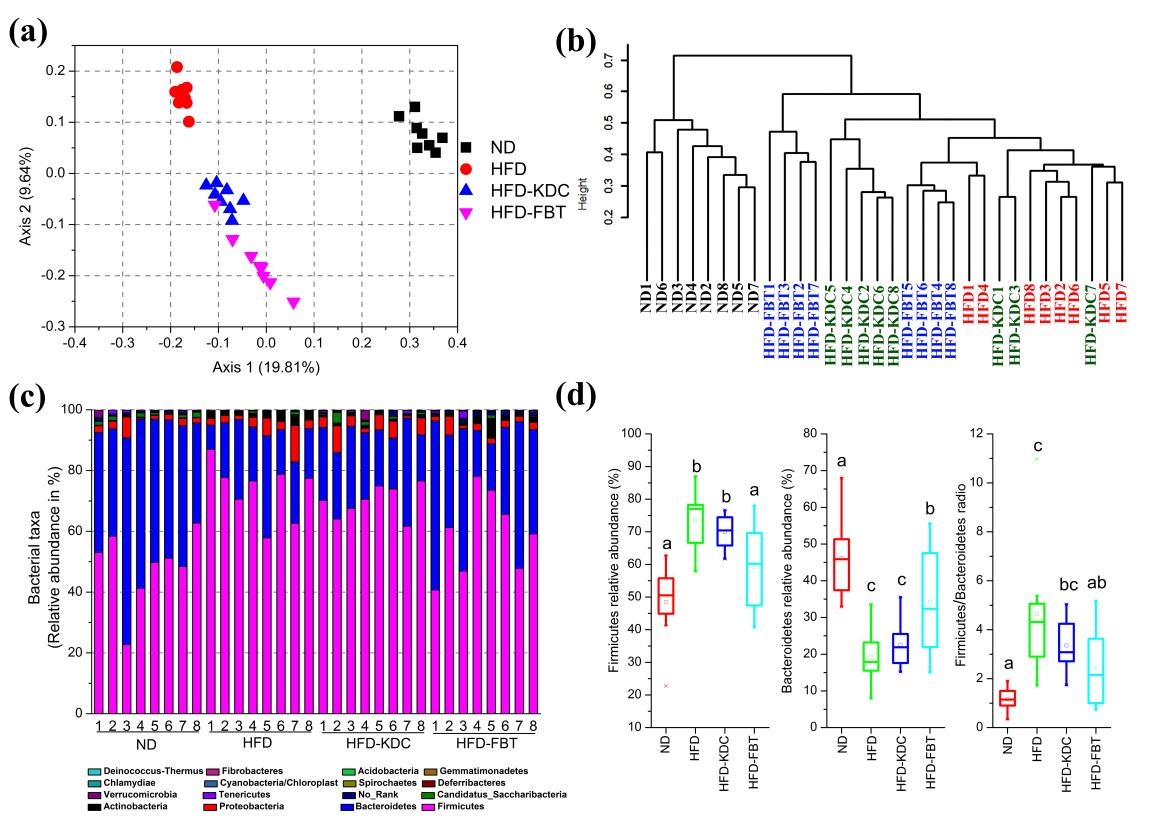
图4 FBT和KDC调节高脂饮食破坏的肠道菌群组成。(a)肠道菌群的PCoA分析;(b)样本UPGMA聚类树;(c)门水平肠道菌群的组成;(d)厚壁菌门,拟杆菌门相对丰度,以及厚壁菌门/拟杆菌门比例。

图5 被FBT或KDC改变的100个OTUs相对丰度。(a)100个OTUs相对丰度热图;(b)被FBT或KDC影响的细菌类群(门、科、属)变化方向。
尽管微生物在冰川地区成土过程和生态系统发展中无处不在,并且起着关键的生态功能,但是细菌和真菌有着截然不同的演替轨迹,并且关于群落的驱动力仍不清楚。在这项研究中,分析了海螺沟冰川变化的七个不同阶段相关细菌和真菌群落,量化其分类组成和演替动态,并从中解读植物建立、土壤发育和线虫对微生物演替轨迹的影响。
观察到的海螺沟冰川衰退始于1823年,自20世纪初以来已明显加速。这项研究是在7个经历长期初级演替的地点进行的,它们分别处于末次冰消期后的不同时间(从裸土到开荒植被群落,最终达到顶极植被群落)。每个阶段的大致年龄根据树的年轮和137 Cs评估的土壤侵蚀速率进行校正,最终分为7个时间阶段(1阶段:冰川衰退后3年,到7阶段:冰川衰退后120年)。
在每个时间阶段,使用3个5×5m的实验地块(在1和2阶段,因为区域较小所以使用2×2m地块)。植物群落的分类确定到种水平,以评估植物的丰富性(包括乔木,灌木和草本层),所有取样的植物材料按种类进行分类,然后烘干和称重。用一个直径为5厘米的土壤取样器从每块地中心和5个角落采集土壤样本,五份土壤混成一份样本,通过2mm筛除去根,约200克的土壤分为三个部分:(1)土壤理化性质分析;(2)土壤线虫群落分析;(3)土壤微生物生物量测定和DNA提取。
细菌16S rRNA扩增子测序(V4-V5)和真菌ITS1扩增子测序;
优势细菌主要是:变形菌门(44.19%)、酸杆菌门(21.25%)、拟杆菌门(9.11%)、浮霉菌门(4.1%)、放线菌门(3.57%)、绿弯菌门(3.10%)、芽单胞菌门(2.34%)和疣微菌门(2.03%)(图1a)。
优势真菌主要是:子囊菌门(48.14%)、担子菌门(36.84%)、接合菌门(4.13%)(图1b)。分:(1)土壤理化性质分析;(2)土壤线虫群落分析;(3)土壤微生物生物量测定和DNA提取。
NMDS分析能把细菌整体分为三个群:第1阶段为cluster1,第2-5阶段为cluster2,第6-7阶段为cluster3,三个cluster之间没有重叠(图1c)。NMDS分析能把真菌体分为2个群:早期(阶段1-5)和后期(阶段6 -7)(图1d)。与真菌相比,每个时间阶段的细菌聚集的更紧密(图1c和d)。
共生网络表明细菌和真菌群落的复杂性在中间时间阶段达到顶峰,其节点和边缘数量最高(图2)
细菌重叠的节点主要属于Proteobacteria(变形菌门)和Acidobacteria(酸杆菌门),真菌重叠的节点主要属于Ascomycota(子囊菌门)和Basidiomycota(担子菌门)(图2)。
通过冗余分析区分了3个细菌群落和2个真菌群落,在环境因子中,ph、总磷、土壤有机碳、凋落物C/N和植物丰富度与微生物群落密切相关(图3a和b)。
变分分析表明相对于生物因素,土壤性质在确定细菌和真菌群落结构方面更为重要,尤其是在早期阶段1–5, 在最后的两个阶段,随着森林的建立,生物因素以及生物和土壤互作越来越重要(图3c和d)。
结构方程模型表明,环境因素影响排名按以下顺序:1)细菌: 土壤全磷(0.66)、土壤pH值(0.64),土壤SOC(0.44), 植物丰富度(0.36)和线虫放牧(0.25); 2)真菌: SOC(0.62), 食线虫(0.57), pH值(0.52),植物丰富度(0.48)和总磷(0.10)(图3e、f)。
四个生物群的丰富度和生物量表现出类似的反应。植物和线虫的丰富度反应最高,细菌的丰富度反应最低,另一方面,真菌的生物量反应最大,其次是细菌、植物和线虫。大多数生物群在第5阶段达到最大的丰富度,在第6阶段达到最大生物量,然后在后期下降。
随机过程支配细菌和真菌群落的变化,而确定性过程支配着植物群落的形成,相反,在线虫中确定性和随机过程大致相等。在最后两个阶段,决定论对细菌和真菌的重要性增加了,与细菌相比,真菌群落组成有更强烈的随机性驱动力。
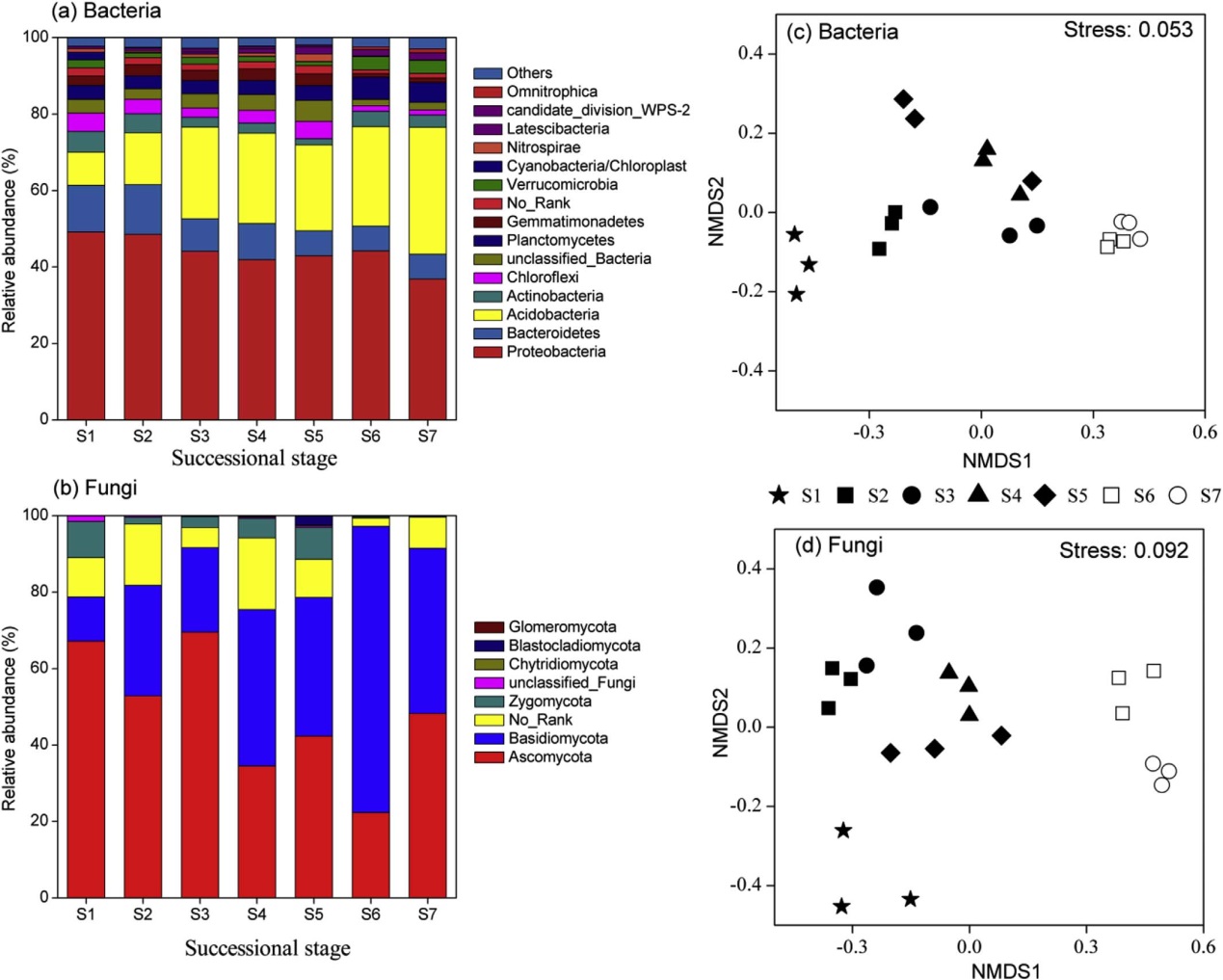
图1 海螺沟冰川不同演替阶段的细菌(A,C)和真菌(B,D)物种组成和NMDS分析。
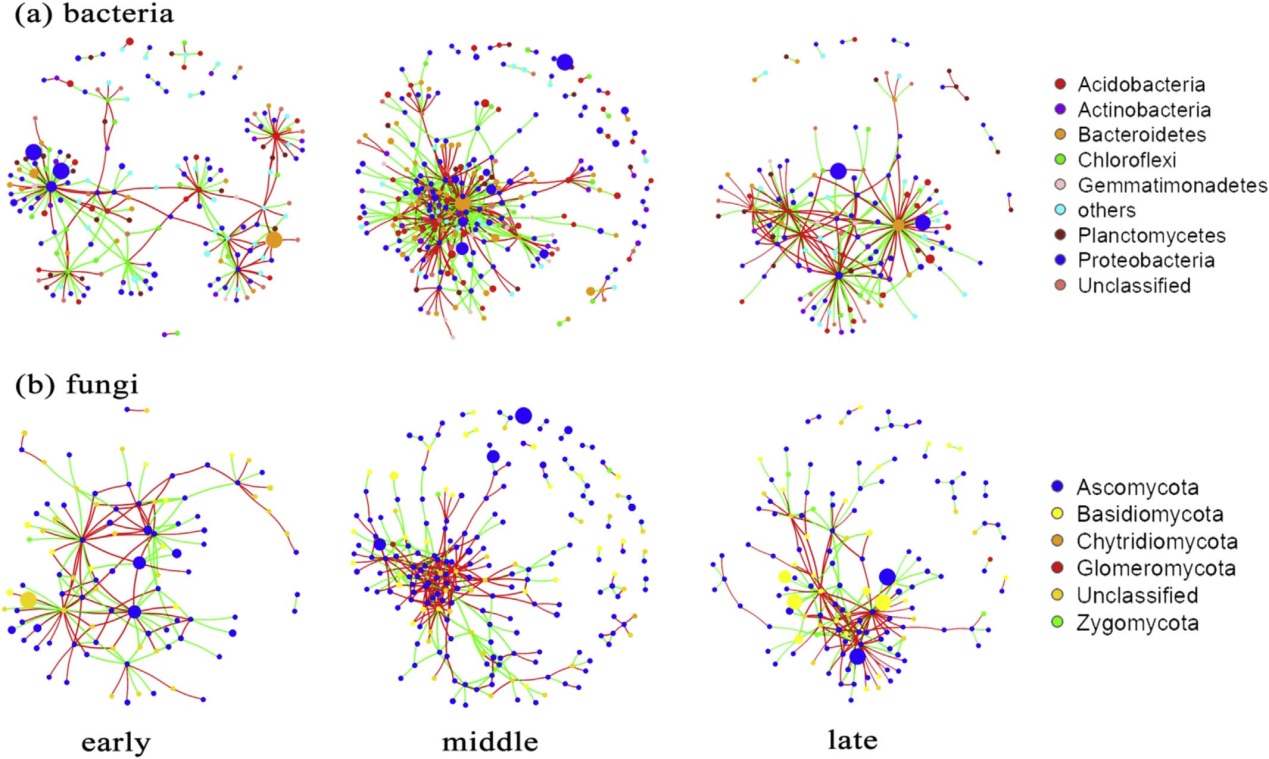
图2 海螺沟冰川不同演替阶段的细菌(A)和真菌(B)共生网络分析。
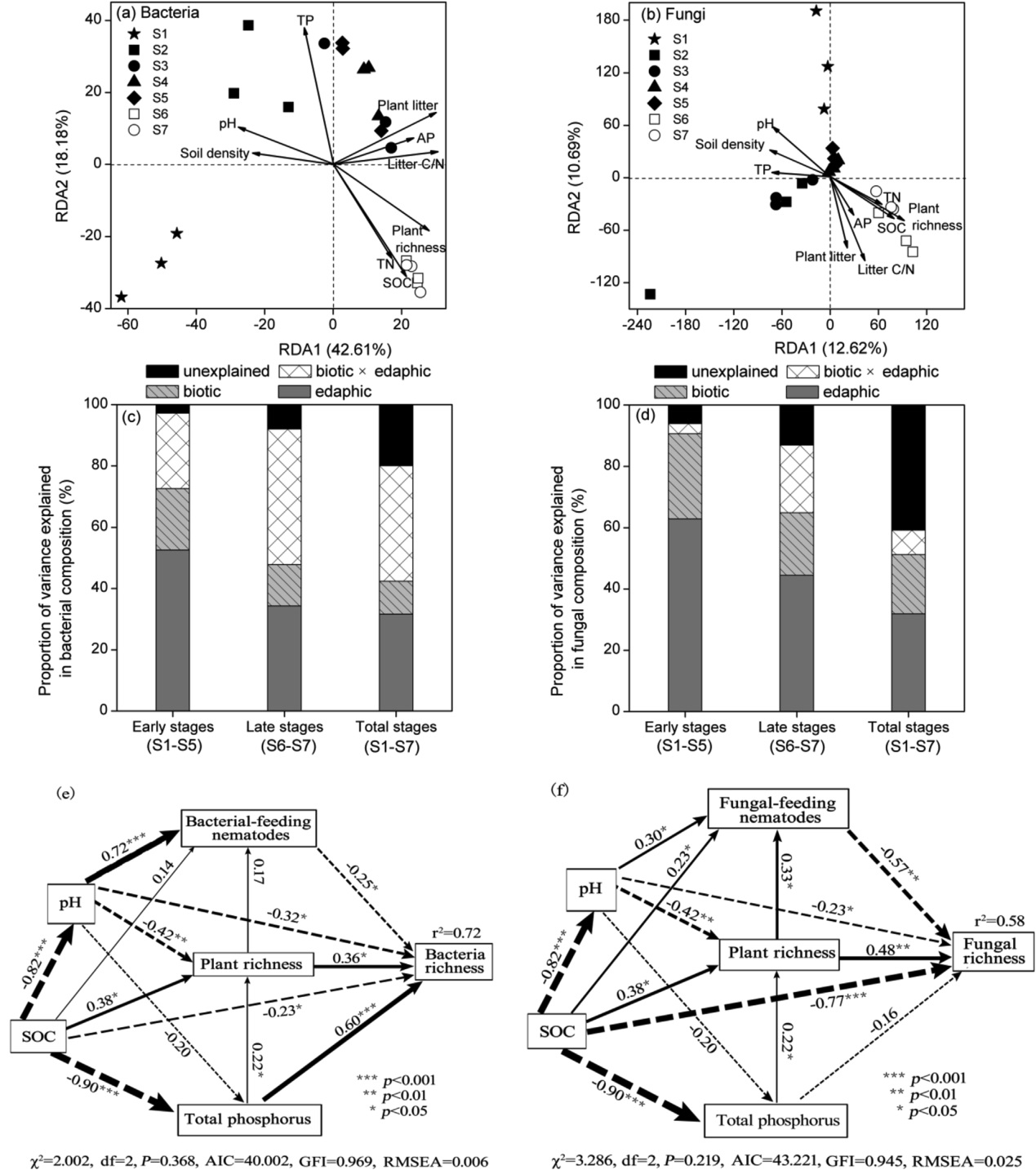
图3 海螺沟冰川不同演替阶段的细菌和真菌群落与环境因素的RDA(a,b)、VPA(c,d)和SEM(e,f)分析。AP速效磷;SOC土壤有机碳;TN总氮;TP总磷。实心箭头和虚线箭头分别表示正相关和负相关,箭头的粗细反映标准化系数大小。

