目的:本研究旨在探讨中国汉族老年人群中常见的R-spondin/Wnt信号基因变异、肠道菌群组成和骨质疏松症 (OP) 风险之间的关系。实验设计:研究者在发现阶段( n = 400, 228 OP 患者)使用目的区域定制靶向捕获二代测序,在验证阶段(n = 768, 356 OP 患者)使用SNPscan®高通量SNP分型技术对参与者进行基因分型检测,并对基因型-OP进行关联分析。利用16S rRNA扩增子测序对与OP相关的肠道菌群情况进行了检测。结果:研究发现LGR6基因中 rs10920362和LGR5基因中rs11178860的遗传变异与 OP 风险显著相关。几个微生物类群与多个骨骼部位的低骨密度(BMD)和 T值相关。调整混杂因素后,rs10920362 与 BMD 相关微生物群之间的关联仍然具有显著性。rs10920362 CT/TT基因型与较低的放线菌、双歧杆菌相对丰度关联。结论:本研究结果表明,LGR6的变异位点可能通过肠道菌群修饰与 OP 发病机制相关。宿主遗传学和肠道微生物组之间的关系为 OP 的预防和治疗提供了新的视角。
骨质疏松症 (OP) 是一种代谢性骨病,其特征是低骨密度 (BMD)和骨组织的微结构退化。在人体中,骨代谢是一个受宿主遗传因素、环境因素和生活方式影响的严格复杂调控下的动态过程。越来越多的全基因组关联研究 (GWAS) 和 SNP 研究表明,R-spondin/wingless 相关整合位点 (Wnt) 信号通路中的基因与 BMD 减少、OP 和骨折的风险相关。富含亮氨酸的重复序列含有 G 蛋白偶联受体 5/6 (LGR5/6) 是 R-spondin/Wnt 信号通路中 OP 易感基因中研究最彻底的成员。LGR5/6作为 R-spondin 家族的受体,通过增强 Wnt/b-catenin 信号传导来调节骨代谢。除了遗传对 OP 的影响外,来自人类和动物研究的各种数据表明,肠道微生物群在调节骨代谢中起着不可或缺的作用。然而,目前还没有关于宿主遗传学、肠道微生物群和OP风险之间相互关系的信息。因此,确定特定遗传变异与涉及该过程的微生物之间的关联是非常重要的。
本研究在某种程度上试图填补这一知识空白,即 R-spondin/Wnt 信号网络遗传变异通过控制肠道微生物群组成导致 OP 风险。因此,我们采用目的区域定制靶向捕获测序技术、SNPscan®高通量SNP分型技术和16S核糖体RNA(rRNA)基因高通量测序技术,对中国汉族人群LGR5/6基因变体、肠道菌群组成和OP风险进行综合分析。
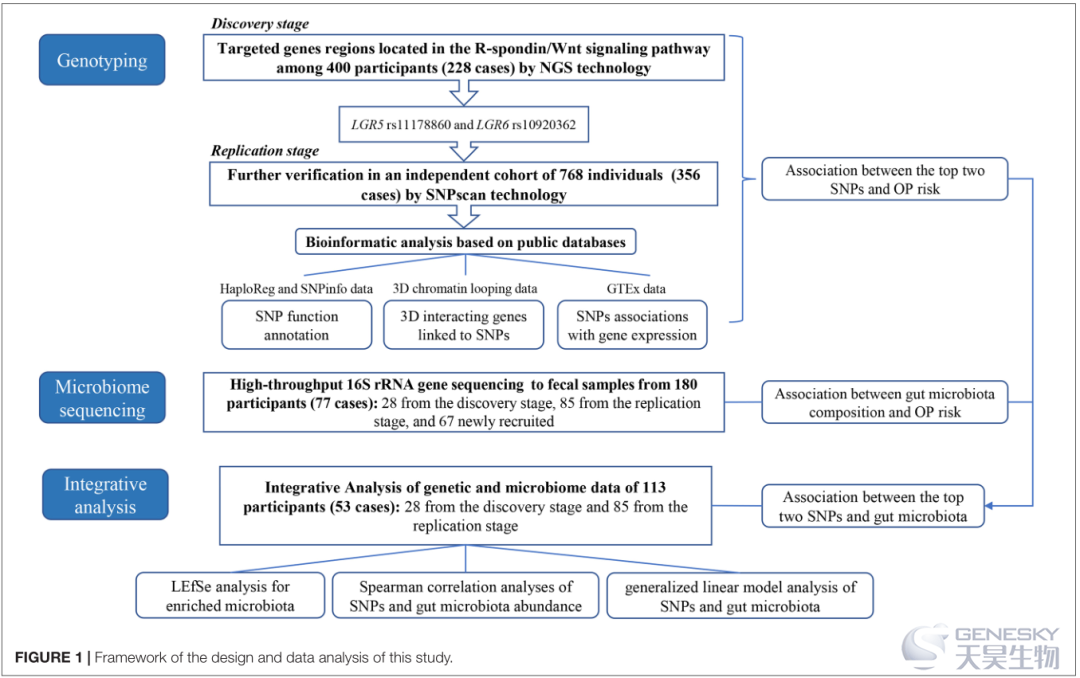
在本研究中,我们首先在 400 名中国参与者(228 名 OP 患者)中对 R-spondin/Wnt 通路基因进行了目的区域定制靶向捕获测序,并使用SNPscan®技术在另一个由 768 名个体(356 名 OP 患者)组成的独立队列中重复了研究结果。然后参考 HaploReg、SNPinfo、3DSNP 和基因型组织表达 (GTEx) 数据进行生物信息学分析,为 OP 基因型关联提供更多证据。之后采用 16S rRNA 扩增子测序技术检测了 OP 相关的肠道菌群变化。最后,结合基因分型和微生物组数据进行联合分析,以阐明 OP 发病机制中潜在的基因组-微生物组关联(图1)。
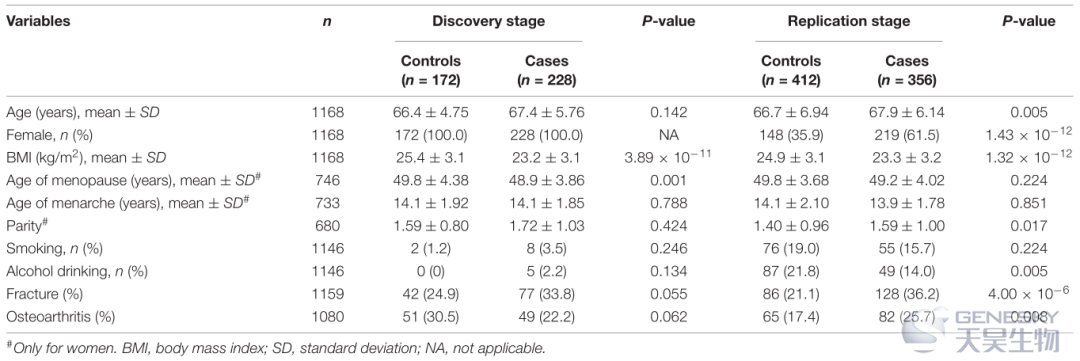
表1为所有参与者的人群特征统计结果。在发现和验证阶段,患有普遍骨质疏松症 (OP) 病例的个体的平均年龄略低于具有正常 BMD 值的对照组。考虑到女性患 OP 患病率高,我们将所有 400 名女性(228 名病例和 172 名对照)纳入基因分型的发现阶段。在 768 个人的验证阶段,我们也包括了男性。
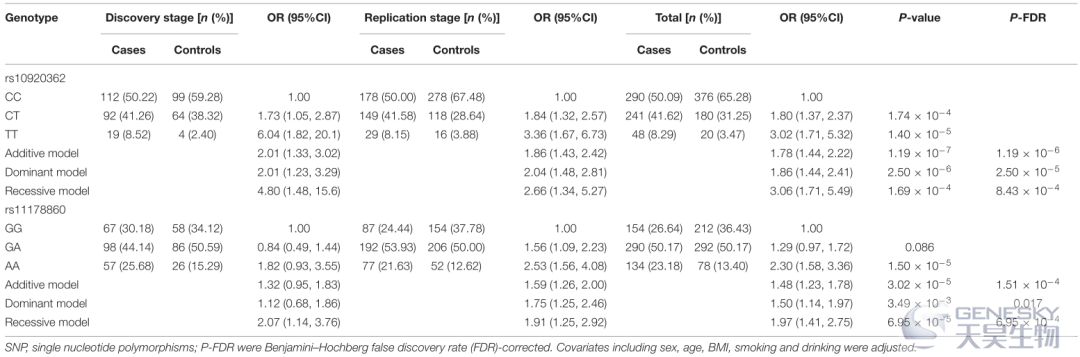
LGR5/6的两种常见变体与骨质疏松症风险之间的关联
本研究采用四种遗传模型(即加性、显性、共显性和隐性模型)来分析SNP 与 OP 风险之间的关联(表2)。在加性模型中观察到,与 C 等位基因携带者相比,rs10920362 T 等位基因携带者患 OP 的风险显著增加。显性模型显示 CT/TT 基因型比 CC 基因型与更大的 OP 风险相关。与 CT/CC 基因型相比,TT 基因型与 OP 患病率风险增加显著相关。至于 rs11178860,与携带 G 等位基因的个体相比,具有 A 等位基因的个体发生 OP 的风险增加。重要的是,显性和隐性模型也显示出结果具有显著性。
表2、前两种常见变异与骨质疏松风险之间的关联表
参考 HaploReg v4.1 的预测,变异 rs11178860 是位于“启动子组蛋白标记”、“增强组蛋白标记”、“基序改变”的各种调节元件中的遗传变异的代理。此外,SNP rs10920362 预计在“DNAse”、“增强组蛋白标记”、“蛋白质结合”、“基序改变”、“GRASP QTL”和“选定的 eQTL”的调节元件中起作用。根据 SNPinfo 的功能分析,rs10920362 被鉴定为“可能具有破坏性”,具有外显子剪接增强子或外显子剪接沉默子的特征。
基于 GTEx 数据集,我们分析了两种遗传变异(rs10920362 和 rs11178860)与各种组织中基因表达的相关性。我们观察到 rs10920362 与甲状腺组织、脑下丘脑、小唾液腺组织中的LGR6基因表达之间的潜在关联。
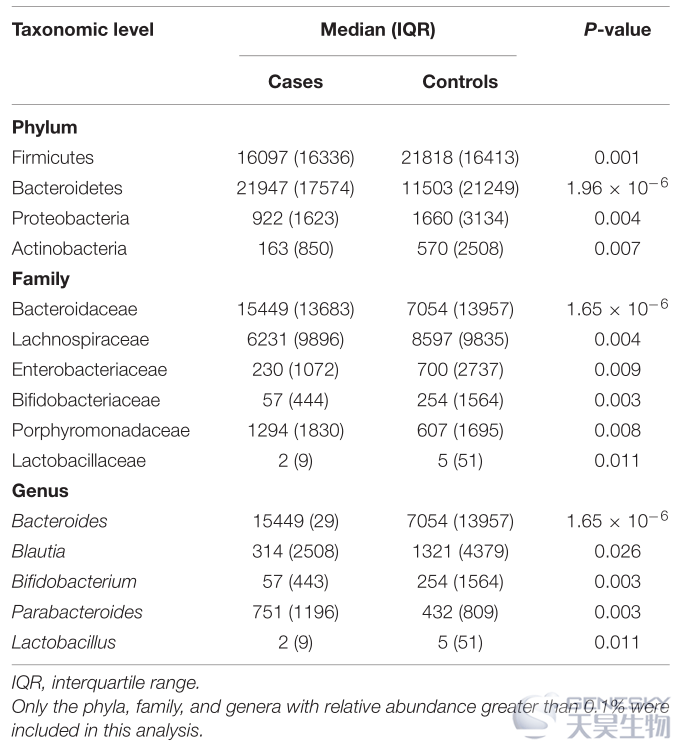
对来自 77 名 OP 患者和 103 名对照者的 180 份粪便样本被纳入肠道微生物群丰度进行分析。结果发现,与对照组相比,OP 组在物种、属、科、目、纲和门水平上的细菌分类群数量相对较少。
在所有样本的肠道微生物群中,厚壁菌门、拟杆菌门、变形菌门和放线菌门主要在门水平上占优势。OP组拟杆菌门的比例显著大于对照组,而厚壁菌门、变形菌门和放线菌门的比例均小于对照组(表3)。Spearman 的相关分析表明,厚壁菌门、变形菌门和放线菌门、双歧杆菌科、乳杆菌科、瘤胃球菌科和瘤胃球菌科以及双歧杆菌、乳杆菌属的水平与所有受试者的 BMD 和T值呈正相关。
表3、骨质疏松症患者和对照组之间在几个分类水平上肠道微生物群组成的比较
这部分共有 113 名参与者(53 名 OP 患者)同时拥有遗传和肠道微生物群数据。共确定了 1,521 个 OTU。维恩图显示 rs10920362 CC 和 CT/TT 携带者共享 973 个 (64.0%) OTU,其中 348 个仅在 CC携带者中发现,200 个仅在 CT/TT 携带者中发现(图 2)。CC携带者的 OTU (1,321) 比 CT/TT 携带者 (1,173) 多。rs11178860 GG 和 GA/AA 基因型共有 864 个 (56.8%) OTU,其中 569 个 OTU 仅在 GG 基因型个体中发现,88 个 OTU 仅在 GA/AA 基因型个体中发现。GG 基因型 (1,433) 中的 OTU 数量大于 GA/AA 基因型 (952)。
LEfSe 分析表明,属于放线菌门、双歧杆菌科和链球菌科的细菌物种在LGR6 rs10920362 CC 基因型中富集。与LGR6 rs10920362 CT/TT 基因型个体相比,CC 基因型个体具有更高水平的双歧杆菌和链球菌(图 3)。
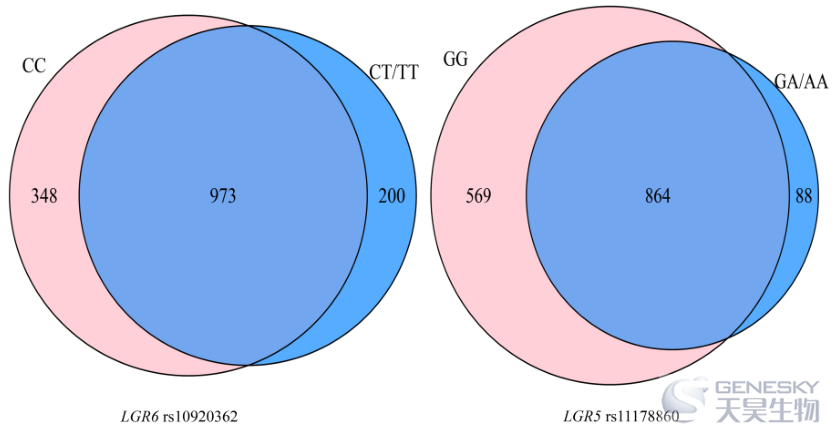
图2、操作分类单位水平上SNPs基因型的肠道微生物群多样性
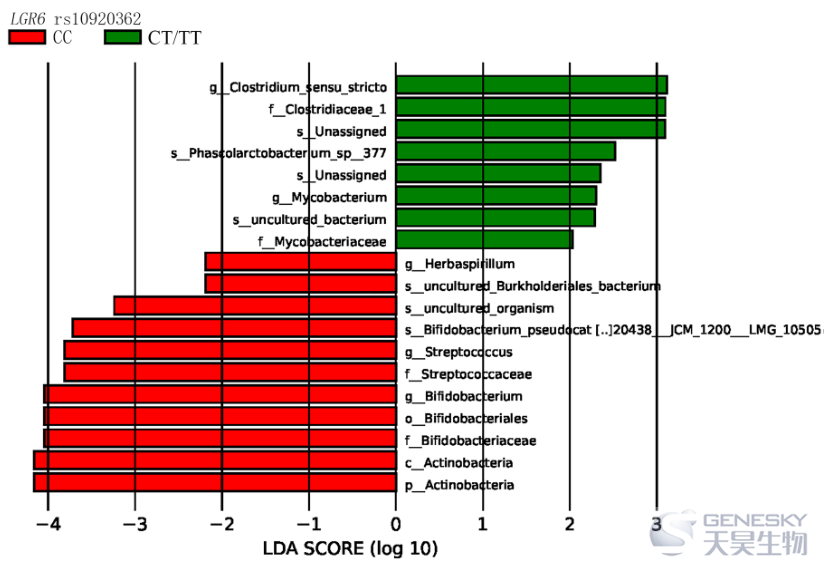
图3、LEfSe显示不同rs10920362基因型细菌类群的差异
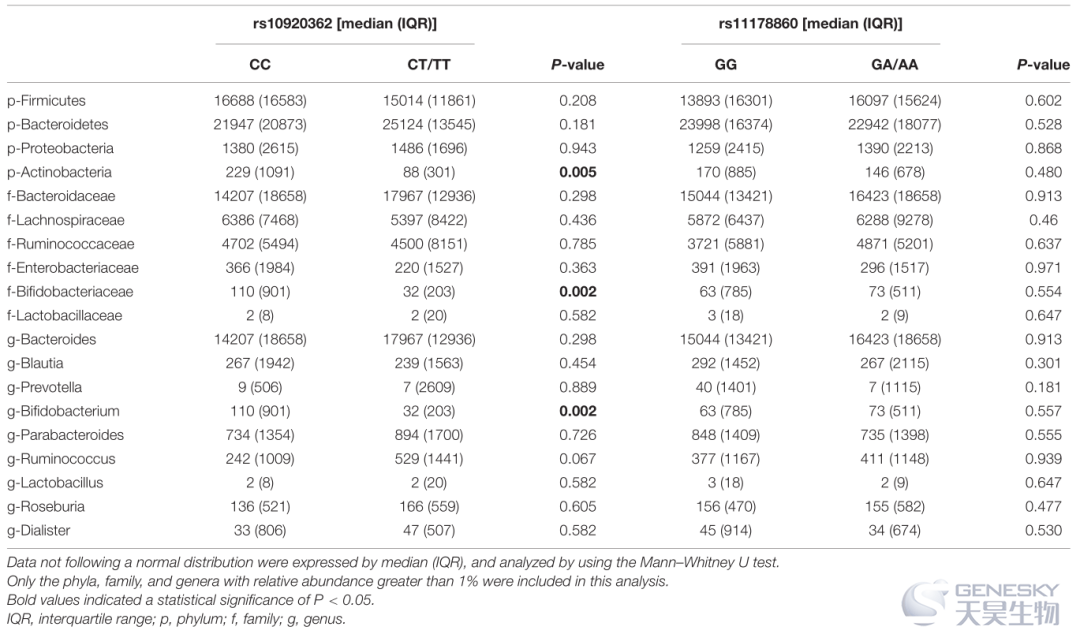
进一步分析的数据显示,rs10920362 CT/TT基因型中放线菌的比例在门水平上显著低于CC基因型(表 4)。在科水平上,携带 CT/TT 基因型的个体的双歧杆菌科丰度低于具有 CC 基因型的个体。在属水平上,CT/TT基因型个体的双歧杆菌丰度低于CC基因型个体。然而,在 rs11178860 GG 和 GA/AA 基因型之间没有发现肠道微生物群落丰度的显著差异。
表4、按rs10920362和rs11178860基因型分层的肠道微生物群组成的比较
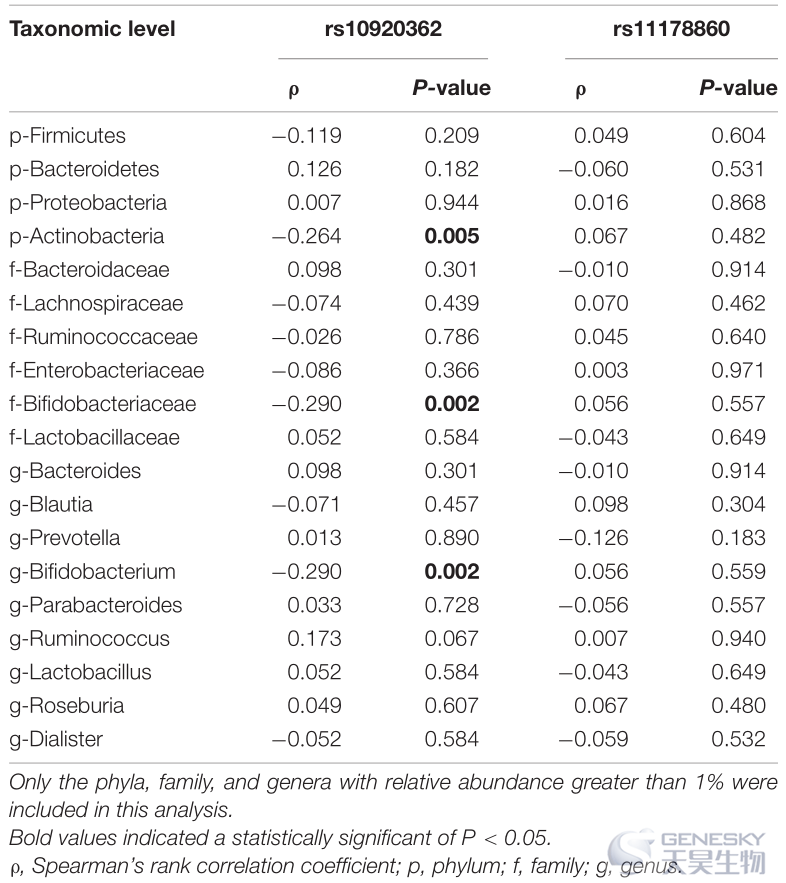
Spearman相关分析显示,放线菌、双歧杆菌和双歧杆菌的丰度与rs10920362 CT/TT基因型呈负相关(表 5)。SNP rs11178860 与肠道微生物群丰度之间未发现显著相关性。
表5、两种常见变异体与肠道微生物群落丰度的Spearman相关分析
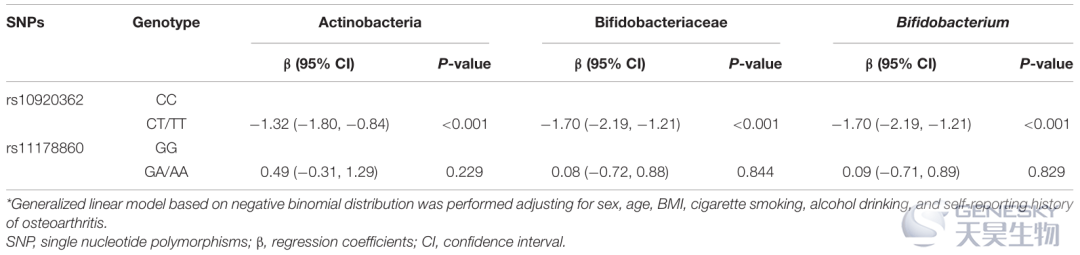
基于上述结果,在广义线性模型中,我们将放线菌、双歧杆菌科和双歧杆菌种的相对丰度作为因变量,rs10920362和rs11178860基因型作为自变量进行分析。与 rs10920362 CC 基因型相比,CT/TT 基因型与较低的放线菌门、双歧杆菌科和双歧杆菌种相对丰度相关,它控制性别、年龄、BMI、吸烟、饮酒和自我报告的骨关节炎病史(表6)。rs11178860 基因型与上述肠道微生物群的相对丰度之间没有发现显著关联。
表6、肠道微生物群丰度对两种遗传变异的依赖性的广义线性模型分析结果
本研究支持LGR5 rs11178860 和LGR6 rs10920362 与 OP 的易感性相关。此外,我们对 R-spondin/Wnt 信号网络基因多态性、肠道微生物群组成和 OP 风险之间关联的综合研究首次表明,LGR6基因中rs10920362 的宿主遗传变异可能会通过降低放线菌门、双歧杆菌科和双歧杆菌种的相对丰度来增加OP 风险。对宿主基因与肠道菌群相互作用的探讨为OP的个体化精准防治提供了新的视角。为了更好地预防和治疗 OP,未来的微生物组靶向治疗应考虑宿主遗传因素。
咨询沟通请联系
18964693703(微信同号)

创新基因科技,成就科学梦想
 咨询热线:400-065-6886
咨询热线:400-065-6886


