eQTLs:能够控制数量性状基因(如身高性状基因)表达水平高低的基因组区域。
sQTLs:能够控制数量性状基因(如身高性状基因)可变剪接水平高低的基因组区域。
英文题目:Regional Variation of Splicing QTLs in Human Brain


研究摘要
人类遗传学的一个主要问题是表达基因的序列变异如何产生组织和细胞特异性分子表型。AS的遗传变异是人类群体中转录组和蛋白质组多样性的普遍来源。本文研究了来自13个人类大脑区域的1209个样本中的sQTLs,使用了来自GTEx项目的RNA-seq和基因型数据。在每个大脑区域都发现了数百个sQTLs。一些sQTLs跨脑区存在,而另一些则显示出区域特异性。这些“区域性普遍存在的”和“区域性特异性的”sQTLs分别显示了SNPs在必需剪接位点内外的不同位置分布,表明它们受不同分子机制的调控。整合结合Motifs和核糖核酸结合蛋白(RBP)与外显子剪接的表达模式,研究发现了可能的因果变异潜在的脑区特异性sQTLs。值得注意的是,SNP rs17651213为剪接因子RBFOX2创造了一个假定的结合位点,并与小脑组织中MAPT外显子3的剪接增加相关,其中RBFOX2高度表达。总的来说,本研究揭示了人类大脑中sQTLs更全面的图谱和区域变异,并证明了这种区域变异可用于精细定位潜在因果变异的sQTLs及其相关神经疾病。

部分研究结果
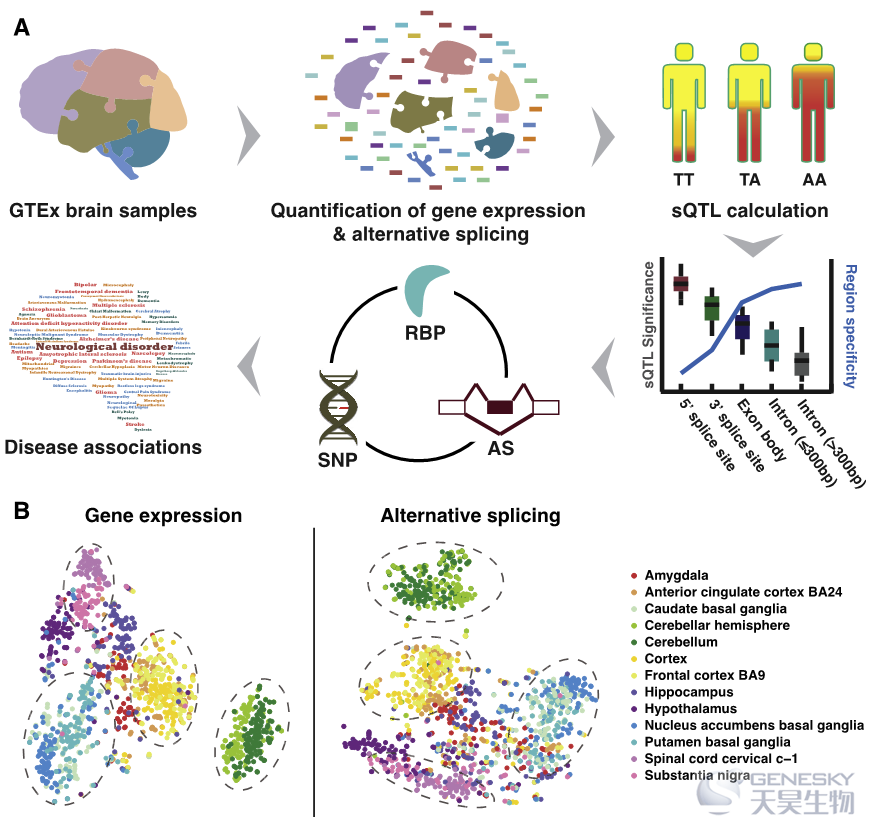
图1、研究和数据概述 (A)研究概述。从人类脑组织样本中获得的RNA-seq数据是从GTEx Portal网站下载的。对数据进行处理,以量化13个大脑区域的基因表达(TPM)和选择性剪接(Percent Spliced In, PSI)。基于选择性剪接和基因型信息,为每个单独的大脑区域鉴定了剪接QTL。sQTL数据用于研究sQTL的位置分布、sQTL的显著性和大脑区域特异性之间的关系。将RBP的表达与AS和SNP数据相结合,以确定潜在的致病顺式变异体和反式调节子。数据被用来确定与14种神经系统疾病潜在相关的sQTLs。(B)基于基因表达(左)和选择性剪接(右)的所有脑组织样本的t-SNE聚类。样本由大脑区域进行颜色编码。虚线椭圆从物理上接近的大脑区域样本。
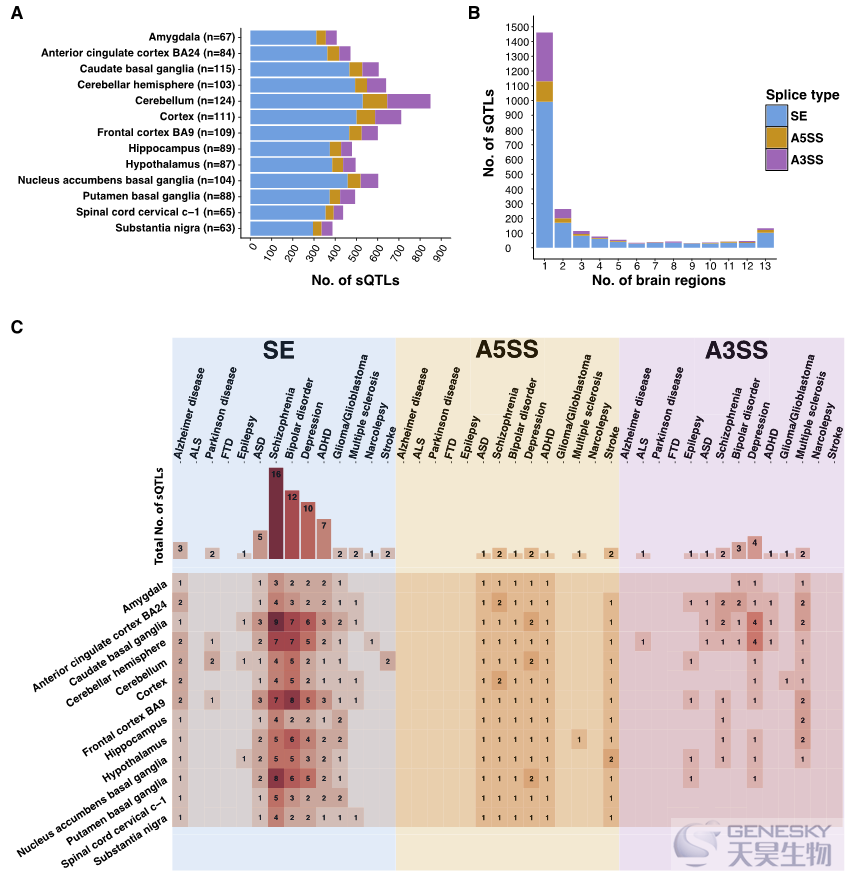
图2、每个大脑区域的sQTLs的识别。(A) sQTLs堆叠条形图,显示在每个大脑区域中识别的sQTLs(包括SE、A5SS和A3SS事件)的数量,括号中的数字指具有可用基因型信息的组织样本的数量。(B) sQTLs堆积柱形图,显示了大脑中被确定为有显著性差异的区域数量的直方图(包括SE、A5SS和A3SS事件)。(C)热图显示了与每个大脑区域的每种神经障碍相关的疾病sQTLs (包括SE、A5SS和A3SS事件)。每行代表一个大脑区域。每一栏代表一种神经类疾病。热图上方的条形图显示了与每种神经疾病相关的独特sQTLs的总数。

图3、利用sQTL信号的区域特异性区分因果sQTL顺式变异体和反式调节子的优先级。(A)顶部:人类MAPT基因的基因结构和六种亚型。在13个外显子中,外显子2、3和10交替剪接,产生6种基因亚型。底部:最长全长MAPT蛋白同种型的功能域(包括外显子2、3和10)。(B)显示小脑MAPT外显子200 kb以内(上)所有SNPs和300 bp以内(下)所有重要SNPs分布图。窗口范围为30 kb(上限)和30 bp(下限)。每个点代表一个SNP,进行颜色注释的为LD最高的sQTL SNP (rs62055489)。y轴显示每个SNP和sQTL外显子之间关联的显著性(log10(p value))。水平线表示显著性截止值(p = 10-5)。(C)气泡图显示了6个SNPs对RBP-RNA结合的影响,正如DeepBind预测的那样。坐标轴显示每个RBP参考等位基因(x轴)或可变等位基因(y轴)序列的RBP结合分数。气泡的大小与两个等位基因间深度结合分数的差异成正比。(D)带有RBFOX结合位点的SNP rs17651213的DeepBind变异图谱。星号表示SNP的位置。(E)显示sQTL (MAPT exon 3和rs17651213)的显著性和每个脑区RBP (RBFOX2)的平均基因表达水平(TPM)的雷达图。(F)显示高LD中的SNP rs17651213(蓝色)和最高sQTL SNP (rs62055489)的LD图(紫色)。(G)组织/细胞类型特异性sQTLs的机理模型图解说明。
英文题目:Identification of eQTLs and sQTLs associated with meat quality in beef
中文题目:牛肉品质关联eQTLs和sQTLs的鉴定
背景:转录具有实质性的遗传控制作用。基因表达的遗传剖析有助于我们理解复杂表型的遗传结构,如牛的肉质。本研究的目的是:1)对背最长肌的肉质性状进行eQTL和sQTL作图分析;2)揭示其表达受近端或远端遗传变异影响的基因;3)识别表达和剪接热点,以及4)揭示影响多个基因表达的基因组区域。结果:选择80头牛进行表型、基因分型和RNA-seq分析。在背最长肌中记录了一组与肉质相关的性状。获得112,042个SNPs的信息,8588个常染色体基因和8467个基因中的87,770个外显子表达数据和剪接数量性状基因座(QTL)图谱(分别为eQTL和sQTL)。先前在该群体中进行的基因、外显子和转录本差异表达分析鉴定了1352个基因,作为与肉品质性状相关的变异性的一部分。eQTL和sQTL定位使用R包矩阵eQTL中的线性回归模型计算的。基因型和出生年份作为固定效应被包括在内,而群体结构是通过将来自基因型数据的主成分分析分析的第一个主成分作为协变量来考虑。用1 Mb作为相关SNP和被分析基因之间的最大距离,将鉴定的QTL分类为顺式或反式。共鉴定出8377个eQTLs,其中反式占75.6%,顺式占10.4%,DEG反式占12.5%,DEG顺式占1.5%;同时发现了11,929个sQTLs,66.1%反式,16.9% DEG反式,14%顺式和3% DEG顺式。共鉴定出27种表达调控因子和13种剪接调控因子,并将其分为膜相关蛋白或细胞骨架蛋白、转录因子或DNA甲基化酶。这些基因可以通过细胞信号或直接转录激活/抑制机制来控制其他基因的表达。结论:在目前的分析中,本研究表明eQTL和sQTL的定位使得鉴定表达调控因子相关基因和转录本成为可能。
研究思路

部分研究结果
在本分析总共发现了11,929个sQTL。最经常识别的sQTL的类型是反式。

图4、利用112,042个SNPs和87,770个外显子(8467个基因)的表达数据,对背最长肌的肉质进行sQTL定位。总共确定了11,929个sQTLs。每个点代表一个sQTL,点大小代表每个关联测试的显著性水平。红色三角形显示了热点的位置。
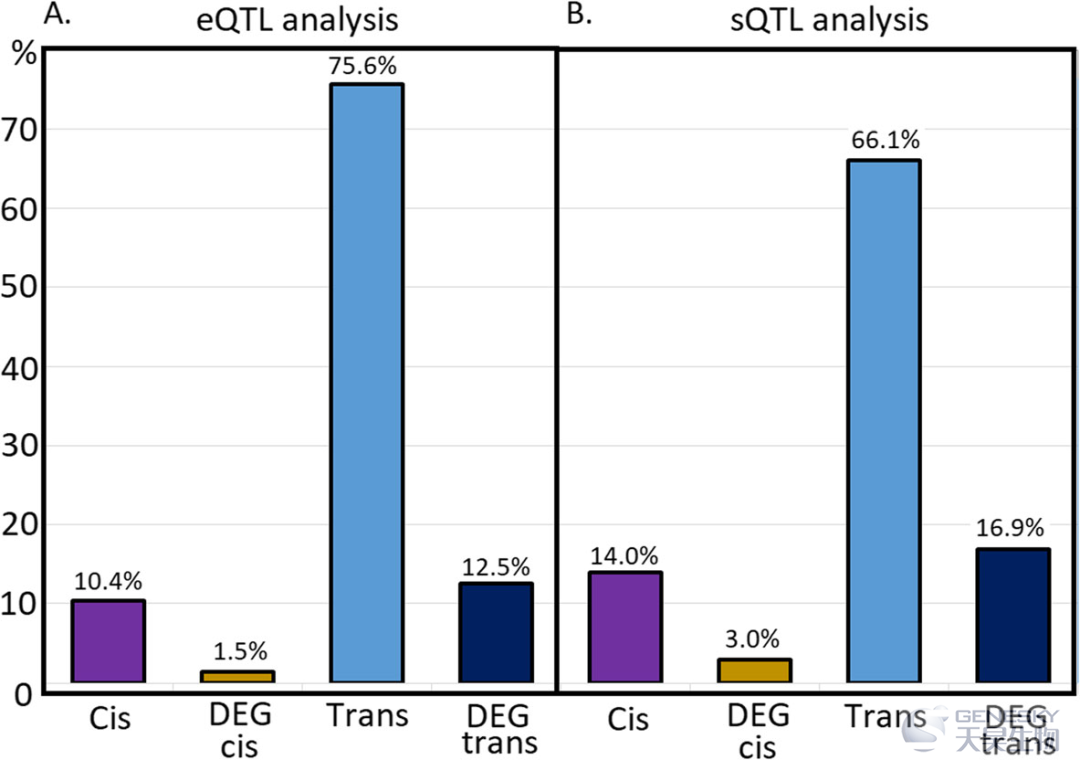
图5、每种eQTL (a)和sQTL (b)的识别频率。背最长肌的肉质相关性状表达QTL作图。
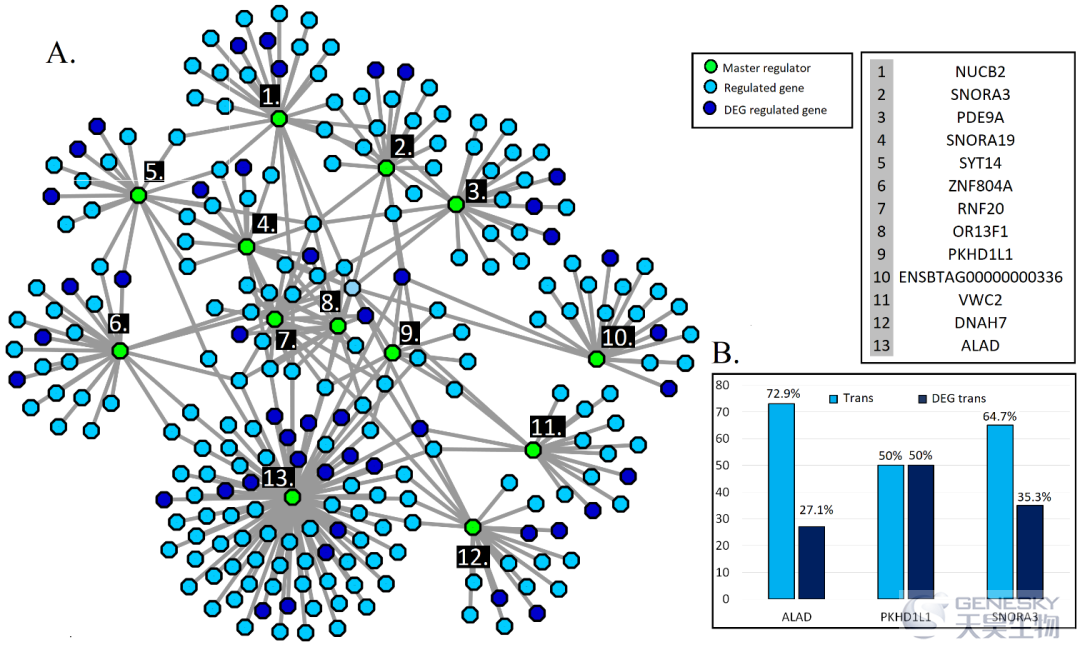
图6、(A)用sQTL作图法鉴定的13个剪接主要调控子和231个调控基因的网络。(B)ALAD、PKHD1L1和SNORA3群中反式和DEG反式调节基因的百分比。13个剪接主要调节子和反式及DEG反式调节基因百分比的网络。
英文题目:Genome-Wide Identification of Splicing Quantitative Trait Loci (sQTLs) in Diverse Ecotypes of Arabidopsis thaliana
中文题目:不同生态型拟南芥剪接数量性状位点的全基因组鉴定
期刊名:Front Plant Sci.
影响因子:4.402
发表时间:2019-10-3
Pre-mRNAs的选择性剪接(AS)有助于转录组的多样性产生,并使植物能够从一个基因产生不同的蛋白质亚型和/或在不同的发育阶段和环境变化中微调基因表达。尽管AS是普遍存在的,但植物中对不同转录本的使用仍是不同的。本研究对666个地理分布多样的拟南芥生态型进行了全基因组分析,以确定可能调节AS差异的基因组区域即sQTLs。尽管sQTLs分布在整个基因组中,但观察到反式sQTL(反式sQTLs热点)在1号染色体上富集。此外,本研究鉴定了几个sQTLs,它们与拟南芥全基因组关联研究中鉴定的性状连锁单核苷酸多态性(SNP)共定位。许多sQTLs富含昼夜节律时钟、开花和应激反应基因,表明在不同生态型的拟南芥中,不同转录本的使用在调节这些重要过程中发挥了作用。总之,目前的研究为影响转录本比率/基因的SNP提供了深入的见解,并促进了对GWAS研究中性状相关SNP的更好的机理理解。据了解这是在拟南芥生态型中进行sQTL分析的第一份报告,可以作为在十字花科中进行sQTL分析的参考。由于全基因组和转录组数据集可用于这些不同的生态类型,因此它可以作为一种强大的资源,用于对性状相关基因座、剪接同工型比率及其表型结果进行生物学解释,以帮助生产高产的作物品种。
研究思路

部分研究结果
表1、利用666种不同生态型的拟南芥绘制的全基因组sQTL总结表。


图7、遗传分析显示了全基因组范围的sQTL分布和1号染色体上反式sQTL的富集。

图8、染色体范围内的sQTLs数目分布图。每组五条染色体(Chr1至Ch5分别用小圆圈和大圆圈表示)在其中一条染色体上显示顺式-sQTLs,在其他染色体上显示反式-sQTLs。
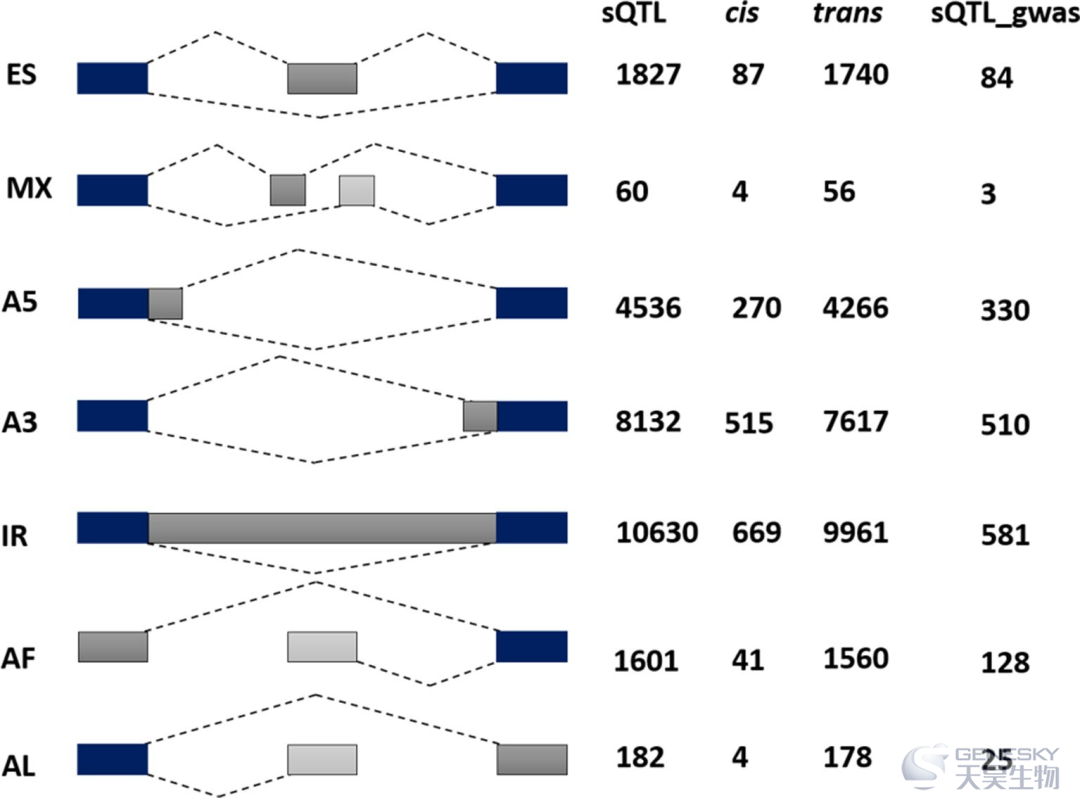
图9、拥有26968个选择性剪接事件的6129个重要的sQTL同源基因的选择性剪接分类。其中1590个与cis相关,25378个与trans类别相关。最后一栏反映了757个sQTL-GWAS同源基因拥有1661个AS事件模式。在剪接变异体类别,包括外显子跳跃(exon skipping, ES)、互斥外显子(mutually exclusive exons, MX)、可变5′/3′剪接位点(alternative 5′/3′ splice-site, A5/A3)、内含子保留(intron retention, IR)、第一/最后可变外显子(alternative first/last exons, AF/AL)中,IR最常见的。

图10、sQTL相关基因的基因富集分析。
天昊生物具有丰富的转录组相关多组学联合分析,以及全转录组测序和qRT-PCR检测经验,我们致力于为研究者提供“一站式”高质量的科研策略咨询、实验技术服务和遗传数据分析服务,期待成为大家科研工作中的“昊”助手与“昊”伙伴。
公司网址:www.geneskybiotech.com
邮箱:techsupport@geneskies.com
往期文章链接:
转录组Plus | 2020年7月研究进展(一):黑色素瘤、抑郁症、CNV疾病、植物不定根发生等;
6个样本全转录组测序,轻松斩获4.2分文章;
全转录组测序在植物研究中的应用(黄瓜、番茄、大白菜、泡桐、拟南芥、茶树、胡杨、玉米);
全转录组测序在动物研究中的应用(小鼠、大鼠、兔、猪、牛、鸡、羊);
mRNA/lncRNA/miRNA综合研究思路解读;
PceRBase:第一个植物ceRNA数据库;
【昊阅读】LncRNA研究,像这个夏天一样“火热异常”;
一套RNA-Seq数据发两篇文章,这篇5分文章可能被你漏掉了;
【昊阅读】RNA-seq揭示油茶冷适应的分子机制;
【昊阅读】Nature:寄生过程“microRNAs制导炮弹”被发现;
【昊综述】环状RNA全方位观察:从生物发生到功能;

创新基因科技,成就科学梦想
 咨询热线:400-065-6886
咨询热线:400-065-6886


前往“发现”-“看一看”浏览“朋友在看”