 咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
今年2月,在Nature Genetics杂志上刊登多篇研究人类肠道菌群与宿主基因组关系的论文,再次让人们把目光聚焦到宿主遗传变异对肠道菌群的影响上。今天我们就分享同期的一篇评论文章,本文以研究宿主遗传对肠道微生物群影响的挑战和未来方向为题,系统概括了该领域取得的成绩及所面临的几大挑战,并强调了系统遗传学(多组学)在研究人类宿主和微生物复杂生态系统中的重要作用。

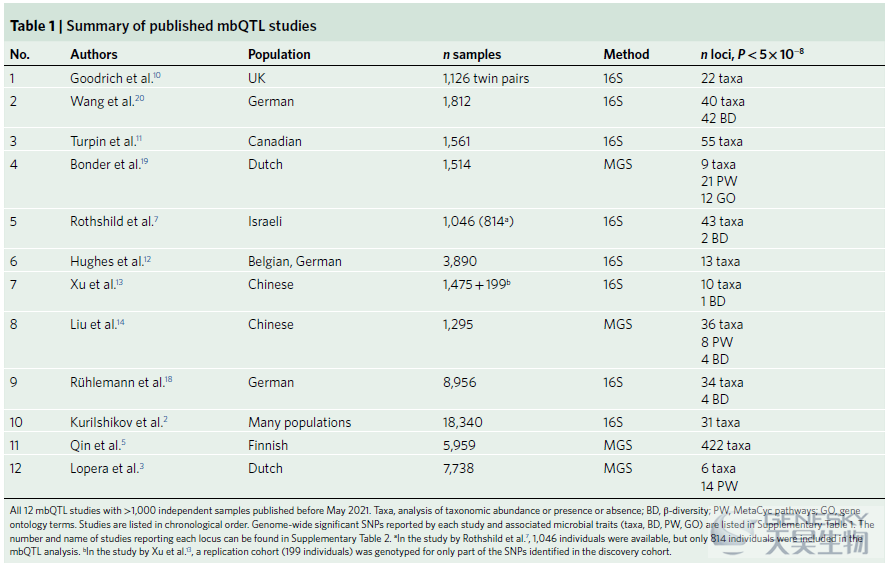
除了LCT和ABO基因座,在12项研究中的至少一项中,P < 5×10-8处还报告了另外546个基因座(图1)。其中,11个位点被两项研究报道,并指向潜在的有趣的候选基因。例子包括CD5,它在T细胞增殖和存活以及其他免疫功能中起作用,以及RBP1,其蛋白产物参与视黄醇(维生素A醇)从肝脏到外周组织的运输。然而,尚不清楚这11个基因座是否代表了真正重复的信号。事实上,在报告它们的两项研究中,没有一个位点被报告与来自同一家族的细菌相关联。
我们期望这些和其他分类群存在遗传信号。MiBioGen联盟和一项荷兰人群研究都确定了显著和提示性命中数与微生物遗传率之间的正相关关系,这表明需要更大的样本量(从而更高的功效)来识别额外的遗传位点。
mbGWAS检测到的大多数全基因组关联的可复制性差是生物现实和正在进行的方法学问题的结果。微生物群落由数百个物种组成,但只有少数存在于几乎所有样本中,许多只存在于一个子集。根据群体群组的估计,大多数分类群将出现在不到50%的样本中,导致遗传分析的有效样本量减半。
群体异质性也可以干扰复制并诱导假阳性或假阴性关联,这是在经典GWAS中首次发现的。例如,FTO基因座与二型糖尿病(T2D)的关联显示出人意料的低重复率。后来发现,FTO变体实际上对体重指数产生影响,而不是直接对T2D产生影响;因此,在体重指数匹配的病例对照队列中,未检测到FTO对T2D的影响。考虑到微生物组受饮食和环境的严重影响,群体异质性也可能在mbGWAS中发挥重要作用,不匹配的队列可能显示较低的复制率。除了这些生物学方面,与样品处理和宏基因组数据处理相关的多种方法问题可能会导致低重复率。
此外,当分析数百或数千个微生物组特征时,经典的全基因组显著性阈值P < 5×10-8可能过于宽松,应考虑考虑多个测试的阈值。事实上,在较大的mbGWAS中,LCT和ABO这两个在队列中不断被重复的基因座是唯一通过这一更严格阈值的基因座,唯一的例外是在芬兰人群中发现的MED13L基因附近的信号。然而,这种变异在非芬兰人群中非常罕见(在非芬兰欧洲人的基因组聚合数据库中,次要等位基因频率为0.0003),因此没有在非芬兰人群中进行测试。
对更严格阈值的要求强调了需要更大的样本量来检测强关联和额外的基因座。这一概念似乎是显而易见的,因为这是我们在GWAS 15年后所学到的。在最初仅在几百个样本中发现具有较大影响的相关基因座后,GWAS迅速转向使用几千个样本的数据集,一些GWAS现在包括超过一百万个个体。与人类数量表型相比,在mbGWAS中,对非常大的样本的需求甚至更迫切,因为大多数分类群存在于< 10%的样本中。然而,对于检测新的mbGWAS基因座所需的最小样本数没有绝对的估计,因为事先不知道它们的效应大小。在这种情况下,一个好的指导方针是在给定的样本量下估计最小可检测效应,这将取决于性状的遗传结构,而不是严格地取决于其遗传率。例如,即使对于高度可遗传的性状,如身高(h2≈0.8-0.9),个体常见变异(如位于HMGA2和GDF5位点的变异)仍然只占总方差的0.3-0.7%,而对于其他具有较低遗传率估计值的性状,如胎儿血红蛋白(h2 ≈ 0.6),已经发现非常大的效应大小(BCL11A基因中的常见变异解释了8-14%)。
以前和最近的mbGWAS的结果可用于估计微生物组性状的最大效应的上限,从而使我们能够推测检测额外基因座所需的样本量。例如,在LCT基因座与青春双歧杆菌的关联解释了0.8%的方差,这意味着至少需要20000个样本来检测解释一半效应(0.4%)基因座的关联。青春双歧杆菌是一个常见的分类单元(存在于> 80%的样本中)。对于不太常见的分类群(患病率为10-50%),需要约30000-135000个样本的数据集来检测类似的遗传效应(图2)。然而,对于其他微生物分类群,可能存在具有更大影响大小的相关遗传变异。例如,对于双歧杆菌科的另一种细菌物种 两歧双歧杆菌,仅在26.3%的样品中存在,发现ABO基因座的遗传变异解释了2.7%的变异。虽然这一估计可能被夸大,或者由于未解释的基因-环境相互作用而被高估,但它确实表明微生物性状背后的多基因结构是复杂的,并且可能在微生物分类群之间有很大差异。
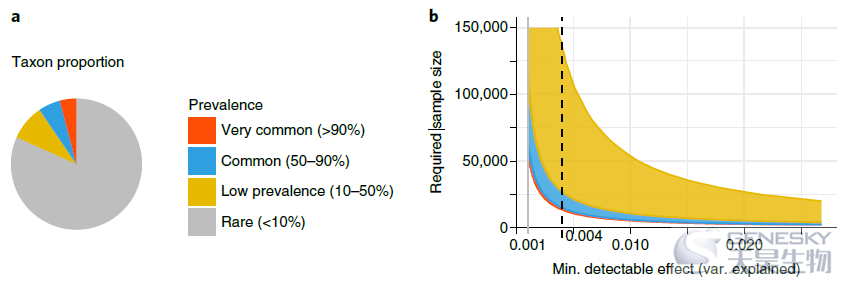
图2、不同分类群流行度的功效分析。
正如我们已经讨论过的,成千上万个微生物样本的样本大小对于识别足够的遗传位置和超越当前建立的基因座是必要的。目前可用的和即将可用的中等规模的MGS队列可以进行荟萃分析,以分析大约20000-30000个样本,前提是分类分类在各研究中协调一致。此外,利用性状间相关性减少表型变异性的多性状GWAS方法也可用于获得功效。
迄今为止,mbGWAS主要关注微生物组成,即微生物分类群的丰度或微生物功能的丰度,例如微生物途径。然而,我们还需要认识到,肠道微生物组的功能不仅可以通过某些物种或代谢途径的丰度来反映,还可以通过微生物基因组中的遗传变异来反映。肠道微生物组包含的基因比人类基因组多100-1000倍,其基因景观可以通过动态适应环境暴露和变化。有证据表明,在全球人口扩张的过程中,肠道微生物群已经与人类基因组共同适应了环境。细菌基因组中可以发生突变,但宿主可能会通过选择压力将细菌的遗传变异保持并传递给下一代。此外,由于通过相同的环境暴露进行的选择,也可以预期人类基因组和肠道微生物组中遗传变异的共现性。
微生物组中遗传变异的关联分析面临几个挑战。首先,肠道微生物组本身的遗传景观在很大程度上仍未被探索,即使这在MGS数据下在技术上是可行的(图3)。主要尝试包括Schloissnig等人的研究,该研究揭示了252份粪便样本中的1030万个SNPs和许多其他类型的遗传变异,以及谢等人的研究,该研究揭示了超过800万个细菌SNPs的分布图,并显示了双胞胎之间较高的相似性,这种相似性在生活了几十年后缓慢降低。最近,Zeevi等人开发了SVfinder管线,并报告了肠道微生物组中超过7000种结构变异。这些研究为探索细菌SNP和结构变异谱奠定了基础。此外,已经开发了几种生物信息学工具来调用来自MGS读序的SNPs,例如metaSNV和inStrain。此外,一般基于概率的SNP鉴定工具,如单倍型鉴定工具,也可用于鉴定细菌SNP,具有极佳的准确性和灵敏性。在未来,长读序测序、单细胞微生物测序和微生物分离物的深度测序将为微生物遗传变异的鉴定提供更高的准确性和分辨率。第二,细菌基因组的突变率通常约为每代0.001,但不同物种之间差异很大。因此,对宿主遗传与细菌遗传变异的相互作用的分析应该集中在暂时稳定的遗传变异上。最近的一项研究评估了338名个体4年来肠道微生物群的遗传稳定性,并确定了几个物种,这些物种在其遗传组成中显示出个体特异性和时间稳定性。微生物遗传组成的个体特异性不仅可以归因于个体的环境暴露,也可以归因于宿主遗传。有趣的是,在生命早期通过母婴传播定居在肠道中的细菌物种随着时间的推移在遗传上是稳定的,并且具有高遗传性,例如双歧杆菌属物种。第三,微生物遗传变异的关联分析,而不是丰度或存在或不存在,带来了优势和挑战。其优势在于以更高的分辨率定义更具体的微生物“表型”,类似于疾病的内表型分析,这可以增强较小样本的功效。缺点是需要进行的统计检验数量增加,这将对检测能力产生负面影响,而检测功效已经是mbGWAS的最大问题。遗传变异体的数量可能在数百万甚至数万亿的规模,远远大于已识别的共同类群和途径的数量。我们设想,随着样本量在短期内可以实现,对微生物基因组使用降维方法(例如,关注单倍型或编码SNP)将是一种新的途径,可以获得对人类遗传-微生物遗传关联的早期见解。
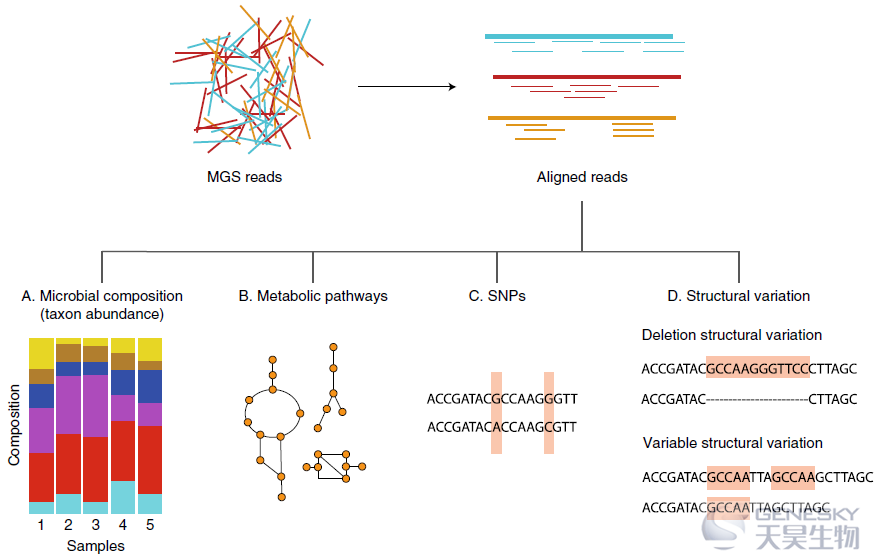
图3:表征MGS的微生物组成和遗传景观。可从MGS数据中获得的关于微生物组组成、功能途径和细菌基因组遗传变异的信息示意图。
值得注意的是,组学方法也已经扩展到微生物领域。例如,宏转录组和宏蛋白质组数据提供了与炎症性肠病和结肠癌相关的细菌途径活性的信息。粪便代谢组学可被视为肠道微生物组的功能读数,主要由肠道微生物组而非宿主基因决定。相比之下,血浆代谢组通常被视为宿主-微生物相互作用的结果。因此,微生物组的组学读数可被视为细菌内表型,以评估宿主遗传对微生物活性和功能的影响。当使用代谢组学数据作为微生物读数时,重要的是要认识到不同的场景面临不同的挑战。首先,将仅由微生物组产生的代谢物与那些受宿主遗传和微生物组控制的代谢物区分开来将是重要的。例如,短链脂肪酸、一些维生素家族和必需氨基酸只能从饮食中获得或由微生物产生。然而,一些代谢物的合成,如次级胆汁酸(BA)和三甲胺N-氧化物,是需要酶参与的,其产生受人类基因组和肠道微生物群的控制。对于这些代谢物,理清遗传学和微生物组的影响是很重要的。例如,在我们最近评估遗传和微生物对BA代谢的影响的研究中,我们使用次级BA与初级BA的比率来校正人类酶的活性。其次,即使对于微生物组驱动的代谢物,它们在粪便或血液中的丰度水平仍然是人类基因组和肠道微生物组的结果,并取决于微生物活性、宿主吸收、运输和消除。例如,95%的短链脂肪酸被结肠细胞吸收并用作能量来源,而只有5%被分泌到粪便中。与这些代谢物的遗传关联可能不仅指向参与微生物活动的基因,而且指向参与代谢物的吸收、使用、运输和消除的基因。第三,当我们将代谢视为微生物读数时,饮食是一个重要的混杂因素。三甲胺N-氧化物和短链脂肪酸的产生在很大程度上取决于肉类和纤维的摄入,但在mbGWAS分析中校正饮食通常很困难,因为在大规模队列中精确测量饮食成分仍然不可行。
除了上述途径,即增加研究样本量、对分类群进行定量分析和关注微生物遗传学,还可以通过研究肠道生态系统中大量未开发的非细菌群落(包括病毒、真菌和原生动物)获得更多见解。尽管肠道中病毒的数量与细菌的数量相似,但病毒体测序的低覆盖率和缺乏用于病毒体分析的通用标记使病毒的研究具有挑战性。可以应用特定病毒体的隔离方案,但是它们很耗时。此外,由于病毒比细菌更具个体特异性,这些分析需要比mbGWAS更大的样本量。
肠道微生物组的遗传学仍然是一个处于起步阶段的领域,在复杂的人类特征上与早期GWAS有许多相似之处。我们预见过去15年从GWAS学到的经验教训,如需要更大的样本量和使用内表型,将帮助我们快速走向新的发现。在荟萃分析中,数据共享和结合GWAS的合作是人类复杂性状遗传研究取得进展的主要因素。因此,我们鼓励在微生物遗传学领域工作的研究人员接受这些科学实践。
即使样本量非常大,可以想象的是,可以用GWAS解释的宿主遗传对肠道微生物群的总体影响将保持适度,在1-10%的方差范围内。然而,我们相信,确定影响肠道微生物组的其他宿主遗传因素,即使是那些影响很小的因素,也将为复杂的宿主-微生物组相互作用提供重要的见解,并可以为疗法和个性化治疗提供信息。例如,LCT基因座中功能性变异的影响相当小,约为双歧杆菌物种丰度变化的0.8%,然而双歧杆菌物种与许多疾病有关,包括对抗癌治疗的反应。因此,结合基因和微生物组筛选有助于改善个性化治疗和预测药物反应。我们期望更大规模的研究将提供足够的力量来调查罕见变体的作用,从而识别其他功能性变异体,这可能为揭示药物反应异质性或开发旨在调节特定微生物的药物提供线索。
最后,我们期望将系统遗传学(多组学)方法应用于人类基因组和肠道微生物组,包括表达、蛋白质组学、代谢组学和其他组学层的整合,这将是未来更好地理解这一复杂的分类学跨界生态系统的必要过程。
天昊生物微生物测序相关链接:
开工大吉,好文读起!《Microbiome》宏基因组 + RNA-seq + 代谢组联合分析揭示糖尿病患者饮食与菌群及免疫关系;
祝贺!天昊微生物扩增子和基因组学联合分析助力客户骨质疏松研究见刊一区杂志《Frontiers in Microbiology》;
喜讯!天昊生物16S扩增子绝对定量测序项目文章再次登陆《Science of the Total Environment》;
祝贺!天昊客户利用16S扩增子测序研究杏鲍菇多糖发酵特性,文章登陆食品科学领域一区期刊《Food Chemistry》;
祝贺!天昊Accu16S细菌绝对定量助力脱氧海水微生物碳氮循环研究登陆一区《Environment International》;
又一篇!天昊Accu16S细菌绝对定量测序项目文章登陆国际顶级环境工程杂志《Bioresource Technology》;
祝贺!天昊Accu16S细菌绝对定量测序项目登陆顶级环境杂志《Journal of Hazardous Materials》;
又一篇!天昊客户微生物16S扩增子测序再登顶级环境科学杂志《Journal of Hazardous Materials》;

创新基因科技,成就科学梦想

