 咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
咨询热线:400-065-6886
在过去十几年中,因为许多抗生素耐药基因(Antibiotic resistance genes, ARGs)具有了抵抗力,加速了微生物对人类健康的威胁进程。目前关于ARGs对健康风险的评估仍然很复杂。诸如ARGs 在病原体中表达的丰度、横向传播倾向和能力等因素都很重要。本研究对来自不同栖息地(6种栖息地,4572样本)的宏基因组测序数据分析,检测到了2561个ARG,它们对24类抗生素具有了抗性。我们通过整合人类可及性、流动性、致病性和临床可用性,对这2561种ARGs对人类的健康风险进行了定量评估。本研究的结果表明,23.78%的ARG会带来健康风险,尤其是那些具有多重耐药性的。文章还计算了四个主要生境中所有样本的抗生素耐药性风险,并通过机器学习成功地绘制了全球海洋生境中抗生素耐药性威胁地图。本研究定量监测ARGs健康风险的新方法将有助于管理这一对人类和动物健康的非常重要威胁。
背景介绍
抗生素耐药性是对人类健康和疾病临床治疗日益严重的全球性威胁。在过去十年中,已在所有环境中检测到抗生素抗性基因(ARG),包括自然、工程和临床及栖息地。包括抗生素的临床使用在内的人为活动,被广泛认为是ARGs传播的主要驱动力。
然而,ARG并非来自当前的人类活动。它们存在于抗生素时代之前,已在永久冻土和古人类粪便中检测到。相反,人为活动推动了从环境和细胞来源中选择基因,这些基因随后可以被用来赋予抗生素抗性。这些基因最初具有一系列环境功能,例如从磷酸盐中释放磷或编码外排泵。不同的基因可能会导致对抗生素的耐药性,因此我们需要确定生物信息学上被鉴定为ARG的基因是否对人类健康构成健康风险,并从多个角度进行评估。
研究方法
研究结果
我们使用一组4572个宏基因组样本来说明ARG分布的全局模式。这些样本是从六种类型的栖息地收集的:空气、水生、陆地、工程、人类和其他宿主(图1)。从这些样本中,我们根据综合抗生素研究数据库(CARD)确定了总共2561个ARG,它们对24种药物类别的抗生素产生耐药性。其中,2401个是仅对一种药物类别产生耐药性的基因,160个是对多种药物类别产生耐药性的基因。在超过75%的样本中发现了25个ARG。另一方面,近一半的2561个ARGs通常由不同的栖息地共享,尤其是赋予对广泛使用的抗生素耐药性的基因,如氨基糖苷类、四环素类和β-内酰胺类。这些结果表明,人为活动,如抗生素的使用,对于ARGs在全球的传播至关重要。
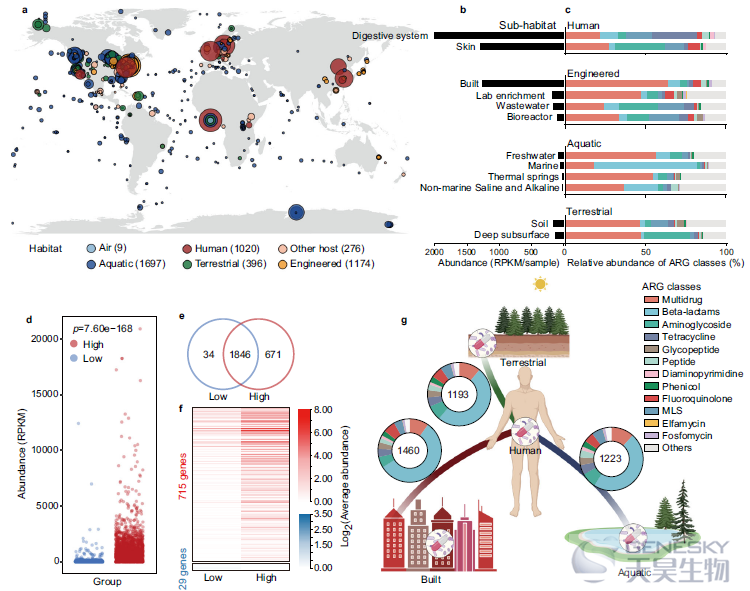
图1、全球抗生素抗性基因(ARGs)的分布模式。a)各种栖息地中富含ARG的样品的地理分布。每个点表示一个采样位置,点的大小反映样本的数量,点的颜色表示栖息地。b)每个亚生境中ARG的丰度。人类的消化系统,主要包括粪便样本,ARGs的丰度最高。c)每个亚生境中抗生素抗性组的组成。仅显示了包含至少 20 个样本的子栖息地。d)高强度的人类活动显着促进了ARGs的丰度。每个点代表一个样本。。e)在人类活动强度低或强度高的地区特定或共享的ARG数量。在高强度人类活动环境中特异性检测到的ARGs有671个。f)在高强度和低强度人类活动环境中,715和29种ARG的丰度分别显着增加。g) ARGs 在与人类相关的三个主要栖息地之间共享。圆圈中的数字表示共享 ARG 的数量。
采样点根据其总体人口密度分为两组,一组活动强度高(>58人/km2),另一组人口密度低。高强度活动区域的ARG总丰度和赋予特定类别抗性的基因显着更高。研究结果还发现,不同的ARGs与人为活动表现出不同程度的相关性,这将影响ARGs对人类生命的健康风险。然后,我们在以下部分中考虑了四个指标(人类可及性、流动性、人类致病性和临床可用性),对每个ARGs的健康风险进行了定量评估。
我们首先检查了人类与其他三个主要栖息地之间共享的ARG,以研究ARG对人类微生物群的可及性。正如预期的那样,建筑环境与人类栖息地(1460)共享的 ARGs最多,而陆地(1193)和水生(1223)环境的ARGs较少(图 2)。这些 ARG 中的大多数被注释为对多药和β-内酰胺类药物的抗性。然后,我们确定了人类栖息地中每种ARG的平均丰度和流行率,并计算了人类可及性。在人类栖息地中仅检测到2561个ARGs中的1714个。这些结果表明,ARGs的人类可及性是可变的,只有一小部分ARGs表现出对人类的高度可及性并构成潜在风险。
我们试图通过实施严格的质量标准来提高ARGs宿主识别的准确性。我们只考虑了超过10 kb的重叠群中的 ARG,并确保在这些包含ARG的重叠群中发现的任何基因的分类从属关系与集合基因组(MAG)的整体分类一致。随后,7555个MAG被确定为ARG的宿主。个体ARGs的宿主在不同的生境中存在显着差异(图2)。大多数ARGs的宿主(89.61%)特定于一个栖息地,只有少数宿主在两个或三个栖息地共享。人工栖息地和人类相关栖息地中的ARG宿主不如自然栖息地中的宿主多样化(图2),也许是人为活动的选择性压力的结果。
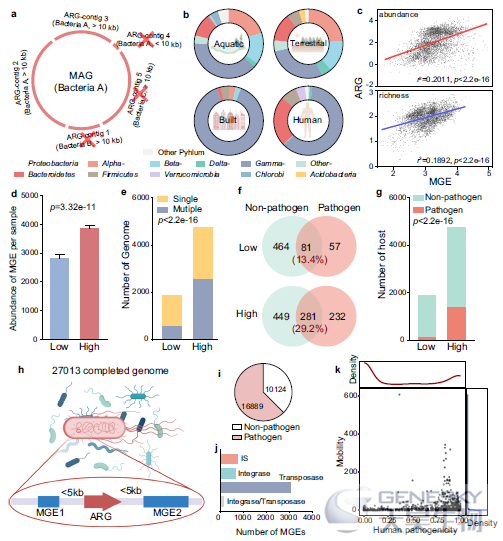
图2、ARGs的人类致病性和流动性。a)ARGs的宿主是通过仅考虑超过10 kb的contigs中的ARGs来确定的,并确保这些包含ARGs的contigs的分类隶属关系与MAG的整体分类一致。b)门水平抗生素抗性宿主的组成显示了四个主要栖息地中ARG宿主的不同分布。c)MGEs在丰度和丰富度上与ARGs呈显着正相关。每个点代表一个样本(n =4572个样本)。ARGs的丰度和数量的值以log10显示。线性回归的结果显示为r2和p值。d)高强度人类活动区的MGEs丰度显着高于低强度人类活动区。e)与低强度人类活动区域相比,高强度人类活动区域中包含多个ARG的基因组数量增加。f)与低强度相比,高强度人类活动区域的病原体或非病原体中的共享ARGs增加。g)与低强度人类活动区域相比,高强度人类活动区域的ARGs致病宿主增加。h)从NCBI RefSeq数据库中收集了27,013个完整的基因组,用于确定ARG的人类致病性和流动性。在所有完整基因组中检测到的ARG上游和下游5 kb用于注释 MGE。i)在完成的27,013个基因组中,有16,889个被识别为病原体基因组。j)我们从完整的基因组中总共鉴定了4612个MGE,其中大部分都涉及转座酶。?)ARGs的人类致病性呈双峰分布,而大多数 ARGs (2266/2561) 的迁移率 <10。
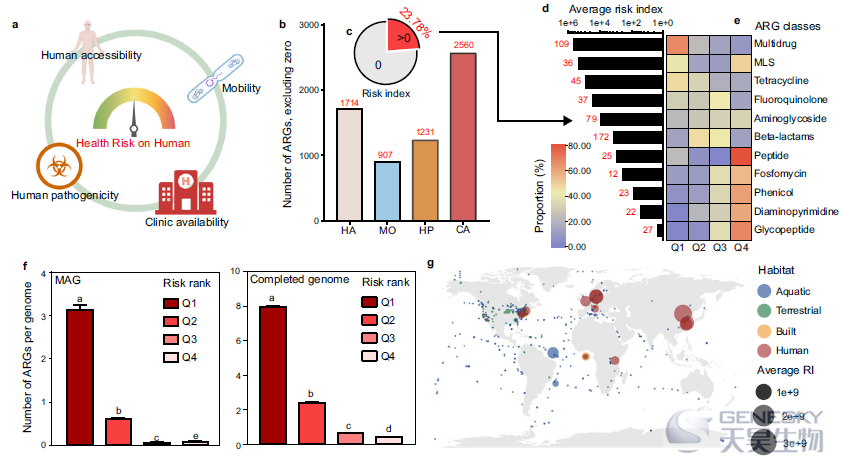
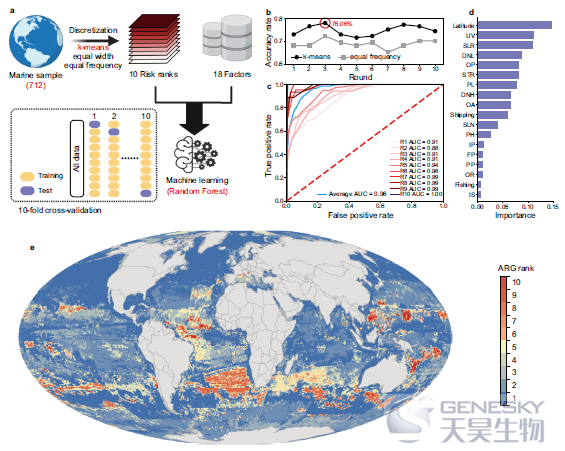
图4、海洋生境中抗生素耐药性威胁的全球图谱。a)机器学习由来自海洋栖息地的712个样本进行训练,并用于预测全球海洋栖息地的抗生素耐药性威胁。b)不同离散化方法的机器学习准确率。K-means表现出比相同频率更高的准确率,并选择最佳模型(准确率=76.06%)进行进一步预测。c)ROC图证实了最佳模型在风险等级分类方面的高性能。d纬度以及气候变化压力源在预测抗生素耐药性风险方面表现出高度重要性。e)由ArcGIS绘制的具有机器学习预测结果的海洋生境ARG风险图。

天昊生物微生物测序相关链接:
开工大吉,好文读起!《Microbiome》宏基因组 + RNA-seq + 代谢组联合分析揭示糖尿病患者饮食与菌群及免疫关系;
祝贺!天昊微生物扩增子和基因组学联合分析助力客户骨质疏松研究见刊一区杂志《Frontiers in Microbiology》;
祝贺!天昊客户利用16S扩增子测序研究杏鲍菇多糖发酵特性,文章登陆食品科学领域一区期刊《Food Chemistry》;

创新基因科技,成就科学梦想

